
はじめに
この記事では、シーケンスリードのクオリティコントロール解析、つまりは品質管理を行うツール群を紹介します。品質管理は、シーケンスデータを解析する前の重要なステップとなります。
データ解析は、クオリティコントロールに初まり、クオリティコントロールに終わる。まさに、柔の精神ですね。
京橋の仙人さん、クオリティコントロールって何ですか?
うむ、まずはデータを知り、柔らかに対応するということじゃ。
前置きはこれくらいで、この記事では、fastqc、MultiQC、Quack、fastpについての基本設定とそれらの使い方を概説します。
実行環境
macOS Big Sur (バージョン11.5.2) MacBook Air (M1, 2020) チップ Apple M1 メモリ 16GB
テストデータのダウンロード
GAGE (Genome Assembly Gold-standard Evaluation)から、 Staphylococcus aureus (黄色ブドウ球菌)のシーケンスデータをダウンロードします。
簡単なサンプル情報は下記に示します。
# テストデータ # Staphylococcus aureus: Data download page # Library 1: Fragment # Avg Read length: 101bp # Insert length: 180bp # # of reads: 1,294,104
まずは、Read1とRead2のFastqファイル(frag_1.fastq.gz と frag_2.fastq.gz)をそれぞれwgetコマンドでダウンロードします。
#ダウンロード wget http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/frag_1.fastq.gz http://gage.cbcb.umd.edu/data/Staphylococcus_aureus/Data.original/frag_2.fastq.gz #ファイルの詳細表示 ls -lh | grep "fastq.gz" #-rw-r--r-- 1 sas staff 61M 4 15 2011 frag_1.fastq.gz #-rw-r--r-- 1 sas staff 65M 4 15 2011 frag_2.fastq.gz
ls -lh | grepでデータサイズを確認したところ、fastq.gz形式で約60Mバイトでした。
それでは、このデータを使って、各ツールでのQCを実施していきます。
関連記事
以下の過去の関連記事です。
Javaの設定: スキップ可です
Mac環境にJavaをインストールします。
Javaのインストールは過去記事を参照ください。
ここでは、パスとバージョンの確認をします。
#パス確認 which java #/usr/bin/java #バージョン確認 java --version #openjdk 18.0.2 2022-07-19 #OpenJDK Runtime Environment Homebrew (build 18.0.2+0) #OpenJDK 64-Bit Server VM Homebrew (build 18.0.2+0, mixed mode, sharing)
fastqcの使い方
Fastqcのインストール
Mac Homebrewでインストールするか、あるいは、 本家のBabraham bioinformatics からバイナリファイルをダウンロードすることができます。
さっき試したところ、本家からのダウンロードは通信が遅過ぎました。 無難に、Homebrewでインストールするのがお勧めです。
Homebrewでのインストールの場合には、
openjdk--19.0.1.arm64_big_sur.bottle.tar.gz
が一緒にインストールされました。
バージョンは、どちらも同じ、FastQC v0.11.9です。
##FastQCインストール #(1) Homebrewでインストール brew install fastqc #(2) Fastqcバイナリファイルを本家からダウンロード wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.zip #(3) Mac用のdmgファイルを本家からダウンロード wget https://www.bioinformatics.babraham.ac.uk/projects/fastqc/fastqc_v0.11.9.dmg
今回は、私自身は(1)のHomebrewでインストールしました。
Fastqcの基本動作
Fastqcをインストールした後、zsh環境で実行しています。
#パス確認 which fastqc #/opt/homebrew/bin/fastqc #バージョン確認 fastqc -v #FastQC v0.11.9 #ヘルプ表示 fastqc -h #出力省略
Fastqcの実行
それでは、いよいよ、テストデータ(fastq.gzファイル)に対して、fastqcを実行してみます。
実行方法としては、個別にファイルを指定する方法と、ワイルドカードでファイルを与えてる方法があります。
#fastq.gzを個別に指定する fastqc --nogroup -o "./" frag_1.fastq.gz frag_2.fastq.gz #ワイルドカード表記も使える fastqc --nogroup -o "./" *.fastq.gz #htmlファイルの表示 ls | grep "fastqc.html" #frag_1_fastqc.html #frag_2_fastqc.html
--nogroupはクオリティスコアの丸め込みを防ぐオプションで、-oの引数では出力フォルダを指定します。
"./"はカレントディレクトリ出力なので省略可能です。
次に、ターミナル上からのhtmlファイルの開き方を説明します。
ブラウザは、Google Chromeか、Firefoxかで開くのがお勧めです(経験上、Safariは非推奨)。
#デフォルト・ブラウザで開く open frag_1_fastqc.html frag_2_fastqc.html #Google Chrome指定で開く open frag_1_fastqc.html frag_2_fastqc.html -a Google\ Chrome #Firefox.app指定で開く open frag_1_fastqc.html frag_2_fastqc.html -a Firefox.app
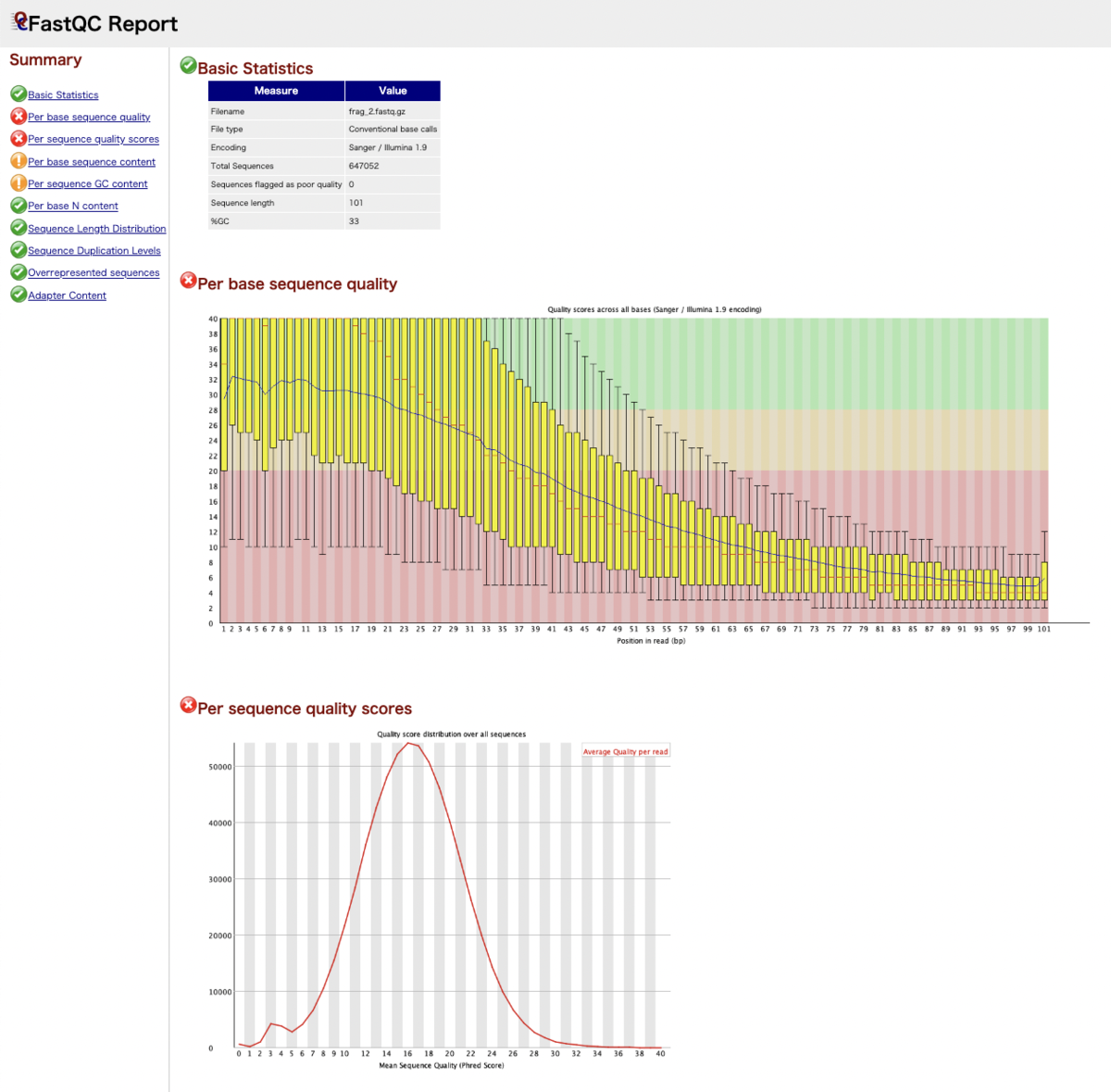
以下のURLで結果レポートをみえます。
frag_1_fastqc.html: https://kumes.github.io/Blog/sh_fastqc/frag_1_fastqc.html
frag_2_fastqc.html: https://kumes.github.io/Blog/sh_fastqc/frag_2_fastqc.html
fastqcレポートの見方
bi.biopapyrus.jpさんのページが分かり易いので、そちらをご参照のこと。
fastqcの原著
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data [Online].
www.bioinformatics.babraham.ac.uk
MultiQCの使い方
次には、複数の分析結果を1つに集約して分析・レポート結果を複合化できる、MultiQCについて紹介します。
例えば、Fastqcコマンド実行後に、生成されたレポートhtmlのあるディレクトリ内でMultiQCを実行します。
MultiQCのインストール
MultiQCは、PyPI(The Python Package Index)のリポジトリから取得するので、 ターミナルを起動して、pipコマンドでインストールします。
#pip3のパス確認 which pip3 #/opt/homebrew/bin/pip3 #PyPIからインストールする pip3 install multiqc #multiqcパス確認 which multiqc #/opt/homebrew/bin/multiqc #multiqcバージョン確認 multiqc --version #multiqc, version 1.13 (759ab2d) #multiqcヘルプ表示 multiqc -h #省略
MultiQCの実行
それでは、MultiQCを実行します。htmlレポートがあるディレクトリに移動して、MultiQCを実行することで、MultiQCのレポートが生成されます。
#fastqcファイルの確認 ls | grep "fastqc.html" #frag_1_fastqc.html #frag_2_fastqc.html #MultiQC実行 multiqc . # /// MultiQC 🔍 | v1.13 (759ab2d) # #| multiqc | Search path : /Users/sas/Desktop/fastqc #| searching | ━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━━ 100% 15/15 #| fastqc | Found 2 reports #| multiqc | Compressing plot data #| multiqc | Report : multiqc_report.html #| multiqc | Data : multiqc_data #| multiqc | MultiQC complete
実行が完了すると、multiqc_report.htmlとmultiqc_dataフォルダが出力されます。
それでは、レポート結果multiqc_report.htmlを開いていみます。
open multiqc_report.html
下図のレポート結果multiqc_report.htmlが表示されます。

MultiQCの原著
MultiQC: Summarize analysis results for multiple tools and samples in a single report. Philip Ewels, Måns Magnusson, Sverker Lundin and Max Käller Bioinformatics (2016) doi: 10.1093/bioinformatics/btw354 PMID: 27312411
quackの使い方
quackは、高速のFASTQ品質評価ツールです。 出力結果は最小限に、実行速度にこだわった開発理念になっています。
quackのインストール
quackは、GitHubからダウンロードします。 binフォルダにquackのバイナリがあり、そのバイナリが使うか、、 もし使えなかったら、ソースからコンパイルしてと記載されていました。
gitコマンドでリポジトリをダウンロードして、quackのバイナリが実行できるかを確認してみます。
quackのバイナリは、Mac版である./quack/bin/MacOS_10.14.6/quackを指定します。
#gitコマンドのパス確認 which git #/usr/bin/git #リポジトリのダウンロード git clone https://github.com/IGBB/quack.git #実行権限の付与 chmod +x ./quack/bin/MacOS_10.14.6/quack #/opt/homebrew/binに移動しておく(パス設定済み) mv ./quack/bin/MacOS_10.14.6/quack /opt/homebrew/bin #ヘルプ表示 quack --help #Usage: quack [OPTION...] #quack -- A FASTQ quality assessment tool # # -1, --forward file.1.fq.gz Forward strand # -2, --reverse file.2.fq.gz Reverse strand # -a, --adapters adapters.fa.gz (Optional) Adapters file # -n, --name NAME (Optional) Display in output # -u, --unpaired unpaired.fq.gz Data (only use with -u) # -?, --help Give this help list # --usage (use alone) # -V, --version Print program version (use alone) #Report bugs to <thrash@igbb.msstate.edu>. #Usage: quack [OPTION...] #Try `quack --help' or `quack --usage' for more information. #もしソースからコンパイルする場合 #cd quack/ #make && make test
quackの実行
Quackでは、入力データとして、gzipされたFASTQ形式のファイル(fastq.gz)を渡して、SVG形式の画像出力を標準出力としています。
片側リードか、ペアエンドリードかで、引数が少し変わってきます。
出力結果のSVGファイルはブラウザ(Google Chrome)で開きます。
#片側リードでの実行(name & adapters の設定なし) quack \ -u frag_1.fastq.gz \ > test_u.svg #ペアエンドリードでの実行(name & adapters の設定なし) quack \ -1 frag_1.fastq.gz \ -2 frag_2.fastq.gz \ > test_p.svg #Google Chrome指定で開く open test_u.svg test_p.svg -a Google\ Chrome

出力結果のSVGファイルには、 片側リードの場合、1つの結果パネルが、 ペアエンドリードの場合、2つの結果パネルが表示されます。
quackのレポートの見方

アレイの各カラムに含まれる各塩基の割合を示す塩基量分布。
各カラムの配列品質分布を示すヒートマップと、アレイ全体の品質スコアの平均値を示すライン
あるスコアに一致する塩基の割合を示すスコア分布グラフで、グラフの左側が100%、右側が0%。スコアの高いデータがグラフの上部に表示されます。
長さ分布グラフ:ある長さのリードが占める割合を示すグラフ
Adapter content distributionグラフ:アレイ中のAdapter contentの分布を示すグラフ
quackの原著
A. Thrash, M. Arick, and D. G. Peterson, “Quack: A quality assurance tool for high throughput sequence data,” Analytical Biochemistry, vol. 548, pp. 38–43, 2018.
fastpの使い方
fastpは、FastQファイルの前処理をオールインワンで高速に行うためのツール群を提供しています。また、C++で開発され、マルチスレッドに対応した高性能ツールでもあります。
fastpでは、フィルタリングやトリミングなども処理実施できるパイプラインも提供されていますが、 この記事では、クオリティコントロールの箇所のみを紹介します。
minicondaの諸設定
まずは、condaを設定するために、minicondaをダウンロード・インストールします。
#Python3.9版 minicondaのダウンロード wget https://repo.anaconda.com/miniconda/Miniconda3-py39_4.12.0-MacOSX-arm64.sh #minicondaのインストール bash ./Miniconda3-py39_4.12.0-MacOSX-arm64.sh -b -p $HOME/miniconda #パス設定を追記 echo 'export PATH="/Users/{Your Name}/miniconda/bin:$PATH"' >> .zshrc cat .zshrc #condaパス確認: HOMEに作成 which conda #/Users/{Your Name}/miniconda/bin/conda #zshの場合、実行?? #source $HOME/miniconda/bin/activate #conda init zsh #設定: 必須ですね conda config --add channels bioconda conda config --env --set subdir osx-64
ややハマったので、このあたりの掲示板を参照しました。arm/M1 Mac版としては配布してないようですね。
fastpのインストール
Minicondaとbiocondaの設定が終わったら、ようやくcondaコマンドで、fastpをインストールします。
#fastpのインストール conda install -c bioconda fastp #パス確認 which fastp #/Users/[Your Home]/miniconda/bin/fastp #ヘルプ表示 fastp --help #省略
fastpの実行
fastpには色々なパイプラインがありますが、クオリティコントロールの方法を紹介します。
トリミングなどを実行しない場合には、入力ファイルとしてfastqあるいはfastq.gzのみを指定する。 フィルタリングはされてQC結果が出力されます。
#fastqファイルの確認 ls | grep "fastq.gz" #frag_1.fastq.gz #frag_2.fastq.gz #片側リードの場合 fastp -i frag_1.fastq.gz -j out1_fastp.json -h out1_fastp.html #ペアエンドリードの場合 fastp -i frag_1.fastq.gz -I frag_2.fastq.gz -j out2_fastp.json -h out2_fastp.html
-jの引数でjsonのファイル名、-hの引数でhtmlのファイル名を指定します。
実行が終わると、html形式のレポートが出力されます。
出力ファイル(-o, -O)をしていないと、処理結果は出力されません。 今回はQCだけなので、指定していません。
下図は、片側リードの結果です。出力結果htmlは各URLから確認できます。

下図は、ペアエンドリードの結果です。
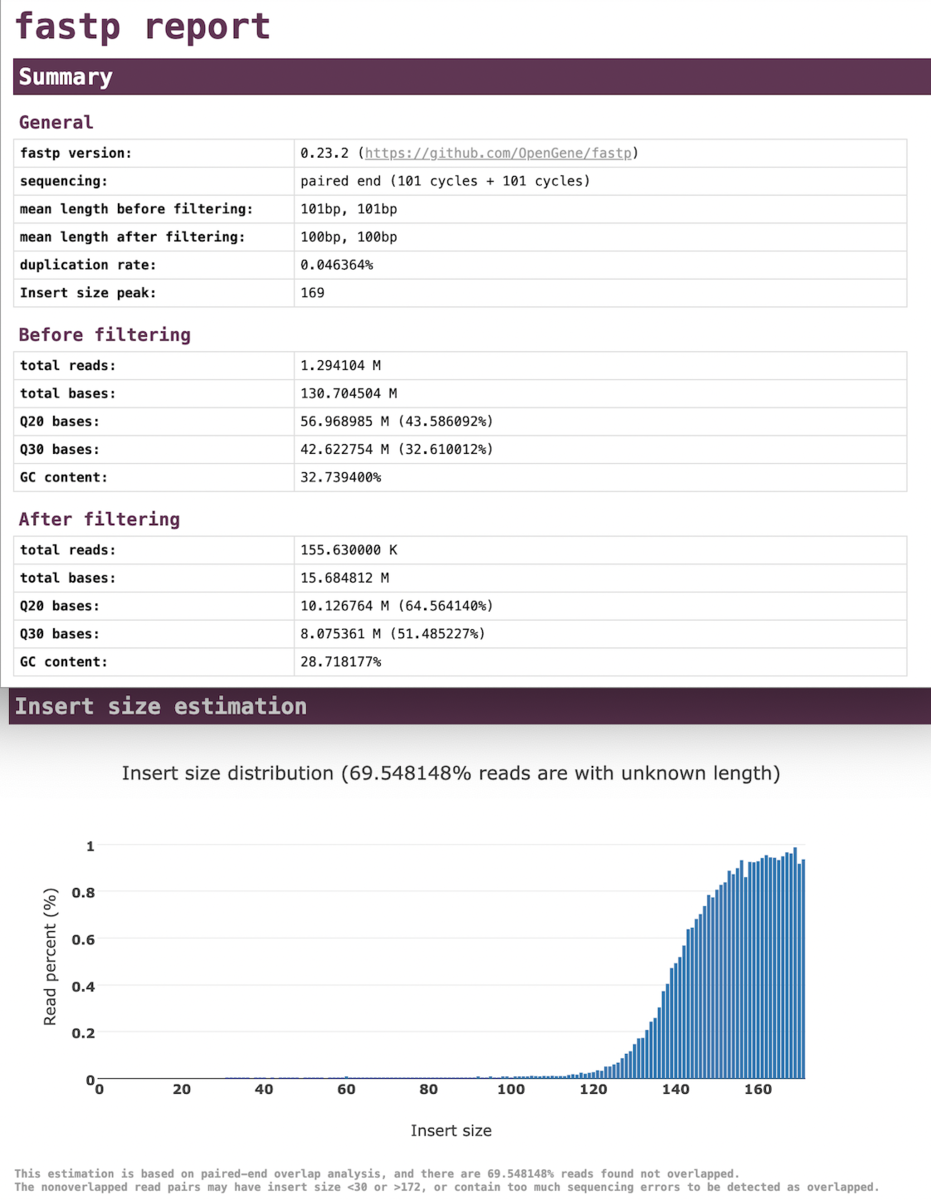
出力結果では、クオリティフィルタ後の結果であったり、インサートサイズの分布を計算されたりと、いい感じの作りになってますね。また、グラフィックが、plotlyで作られているのもオツですね。
やや、htmlレポートが長くて、比較が難しいのが厄介かもですが。
ペアエンドリードでのクオリテイィトリミングの実行例
fastpでは、クオリティフィルタリングの前後でQCレポートを生成します(そのため、htmlレポートがやや長い)。
下記の条件は、 塩基レベルでの品質値30以上(-q 30)、3'末端のクオリテイィフィルタリング(-3)、ReadのN塩基数10未満(-n 10)、read1の末尾1塩基の削除(-t 1 )、read2の末尾1塩基の削除(-T 1)、長さフィルタリング25塩基以上( -l 25 )、3スレッド(-w 3)での実行例です。
#ペアエンドリードの場合 fastp -i frag_1.fastq.gz -I frag_2.fastq.gz -3 -q 30 -n 10 -t 1 -T 1 -l 25 -w 3
fastpのオプションの説明
| オプション | 概要 |
|---|---|
| -i, --in1 | read1の入力ファイル名(文字列[=]) |
| -o, --out1 | read1の出力ファイル名(文字列[=]) |
| -I, --in2 | read2の入力ファイル名(文字列[=]) |
| -O, --out2 | read2の出力ファイル名(文字列[=]) |
| -A, --disable_adapter_trimming | アダプタのトリミングはデフォルトで有効。このオプションが指定された場合、アダプタのトリミングは無効となる。 |
| -a, --adapter_sequence | read1用のアダプタ。SE data: 指定しない場合、アダプタは自動検出される。PE data: R1/R2が重複していないことが判明した場合に使用される。(文字列 [=auto]) |
| --adapter_fasta | FASTAファイルを指定し、read1, read2 (PE)をこのFASTAファイル中の全配列でトリミングする (文字列 [=]) |
| -f, --trim_front1 | read1の先頭何塩基をトリミングするか(デフォルト: 0) (int [=0]) |
| -t, --trim_tail1 | read1の末尾何塩基をトリミングするか(デフォルト: 0) (int [=0]) |
| -F, --trim_front2 | read2の先頭何塩基をトリミングするか(デフォルト: 0) (int [=0]) |
| -T, --trim_tail2 | read2の末尾何塩基をトリミングするか(デフォルト: 0) (int [=0]) |
| -5, --cut_front | 5'末端(先頭)から後方へウィンドウを移動し、その平均品質 < threshold ならウィンドウ内の塩基を削除し、そうでないなら停止する。 |
| -3, --cut_tail | 3'末端(末尾)から前方へウィンドウを移動し、その平均品質 < threshold ならウィンドウ内の塩基を削除し、そうでないなら停止する。 |
| -Q, --disable_quality_filtering | クオリティフィルタリングはデフォルトで有効。このオプションが指定された場合、品質フィルタリングは無効となる。 |
| -q, --qualified_quality_phred | 塩基レベルでの品質値を指定する。デフォルト15で、phred quality >=Q15 でトリミングすることを意味する。(int [=15]) |
| -n, --n_base_limit | あるReadのN塩基数がn_base_limitを超えた場合、そのread/pairは破棄される。デフォルトは5 (int [=5]) |
| -e, --average_qual | あるReadが、平均品質スコア < avg_qual ならば、そのread/pairは破棄される。デフォルト=0は要求しないことを意味する (int [=0]) |
| -L, --disable_length_filtering | 長さフィルタリングはデフォルトで有効。このオプションが指定された場合、長さフィルタリングは無効となる。 |
| -l, --length_required | length_requiredより短い、Readは捨てられる。 デフォルトは15。 |
| -w, --thread | ワーカースレッド数、デフォルトは3 (int [=3]) |
fastpの原著
Shifu Chen, Yanqing Zhou, Yaru Chen, Jia Gu; fastp: an ultra-fast all-in-one FASTQ preprocessor Bioinformatics, Volume 34, Issue 17, 1 September 2018, Pages i884–i890,
まとめ
4つのクオリティチェックのツールをインストールから基本的な使い方を概説しました。
最近は、htmlレポートが標準出力なので、見易いですね。 下流解析から戻って再確認するにも、human-Readableって良いですね。