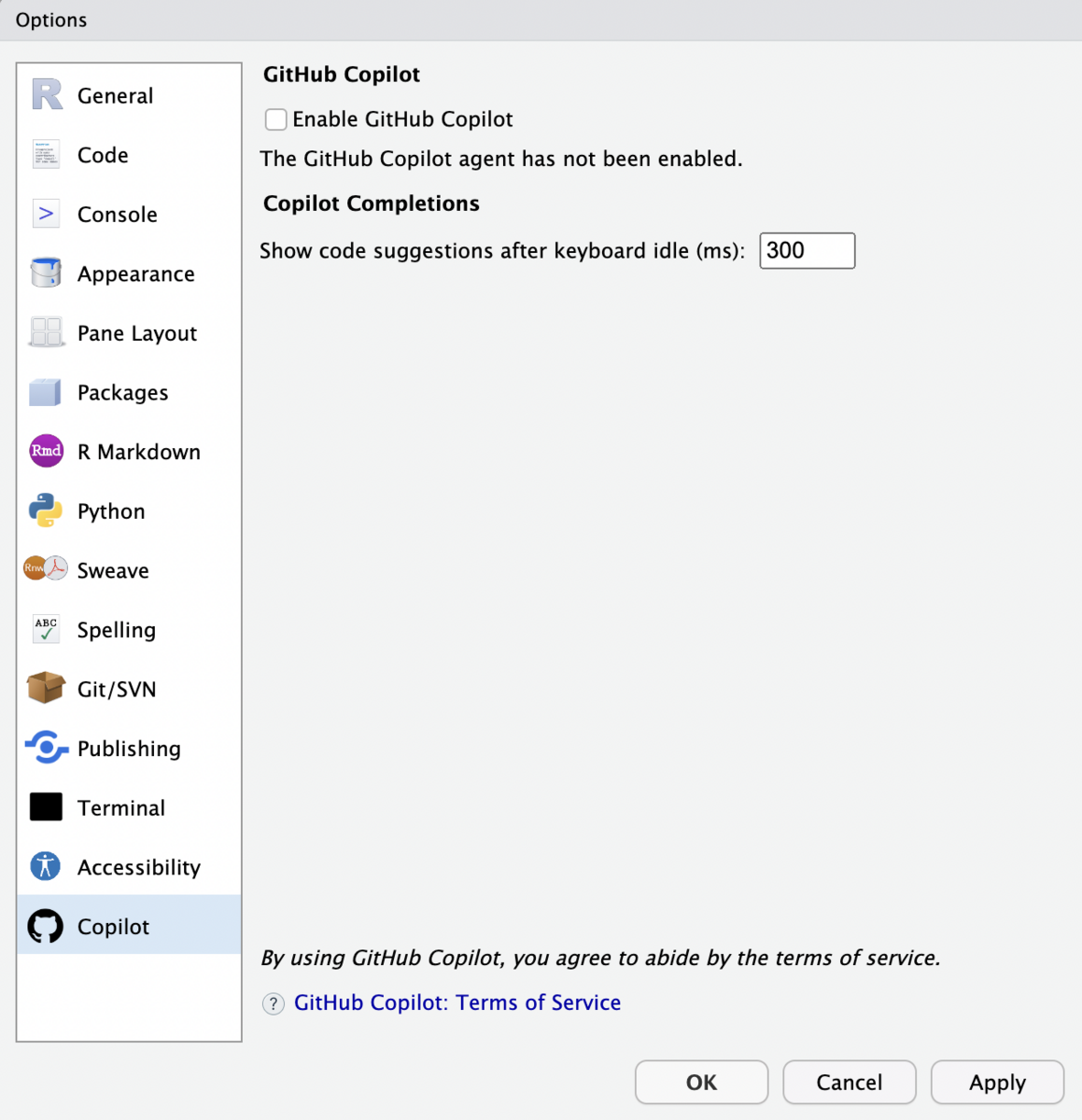
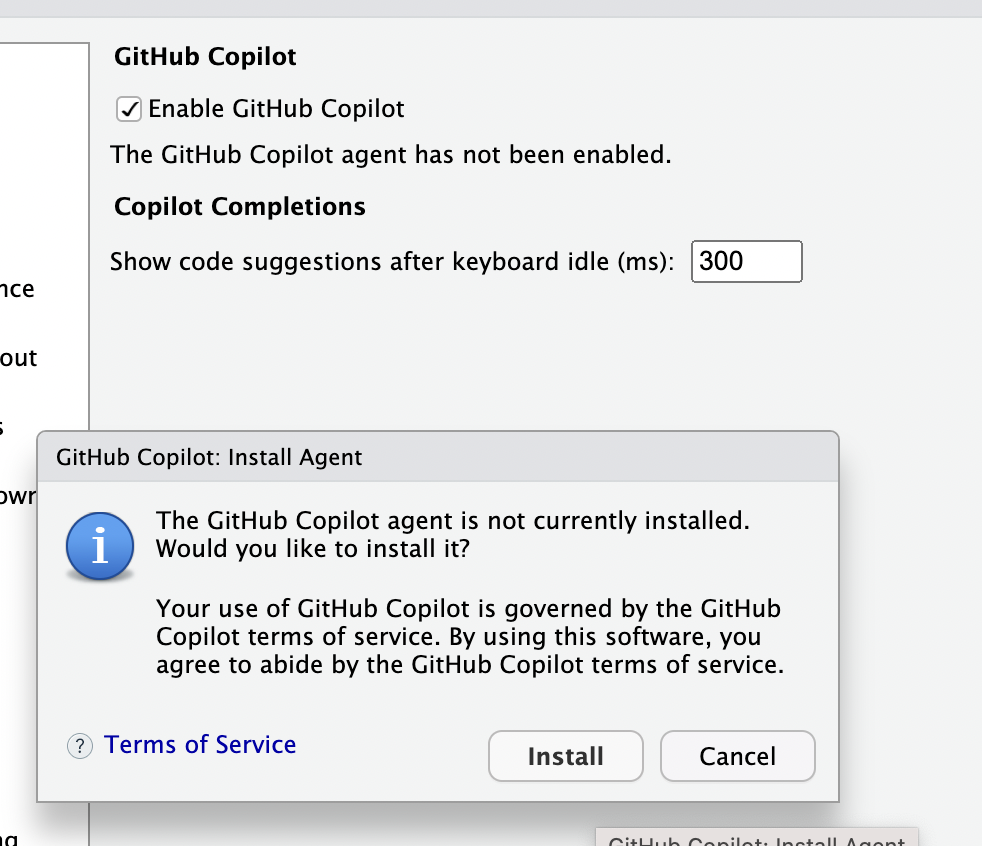
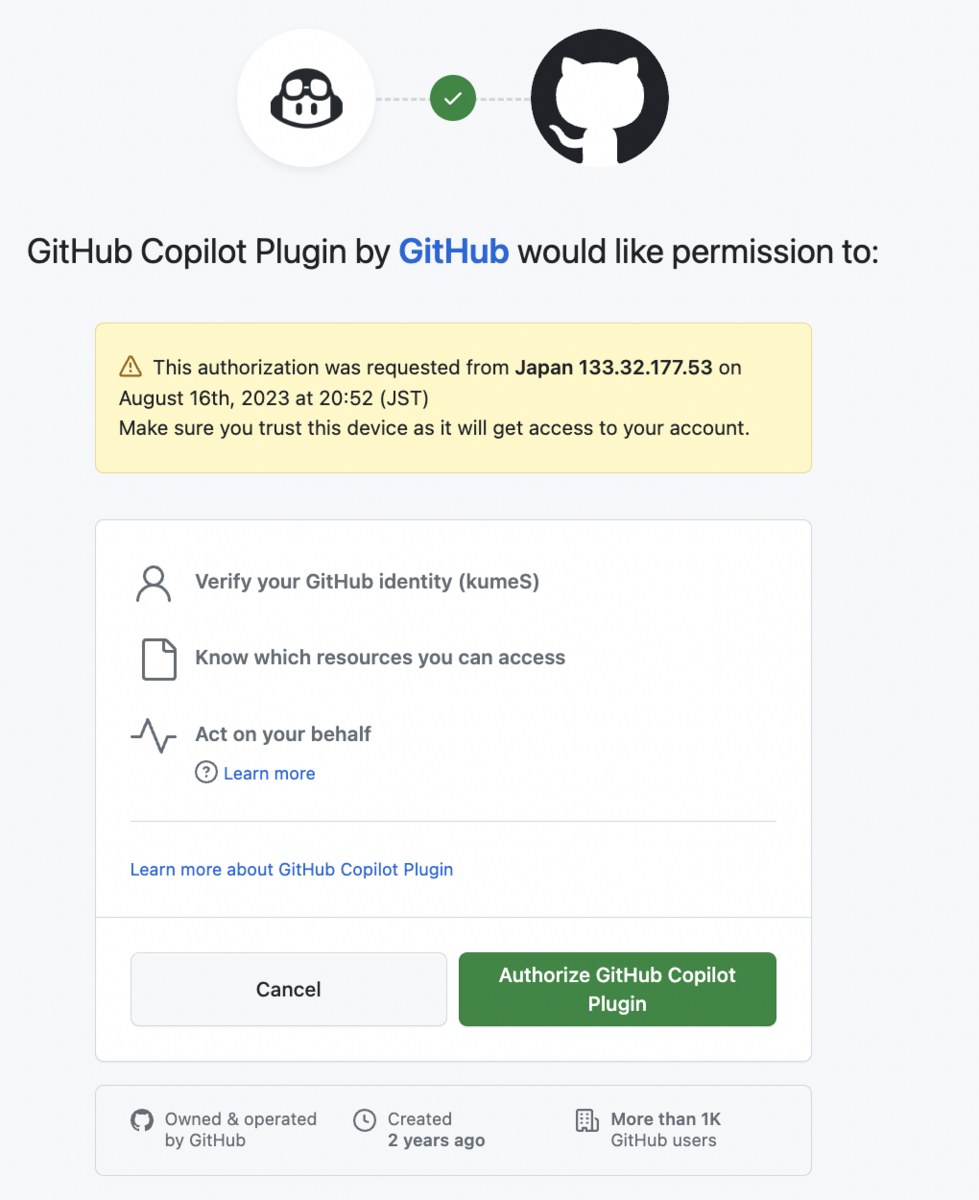
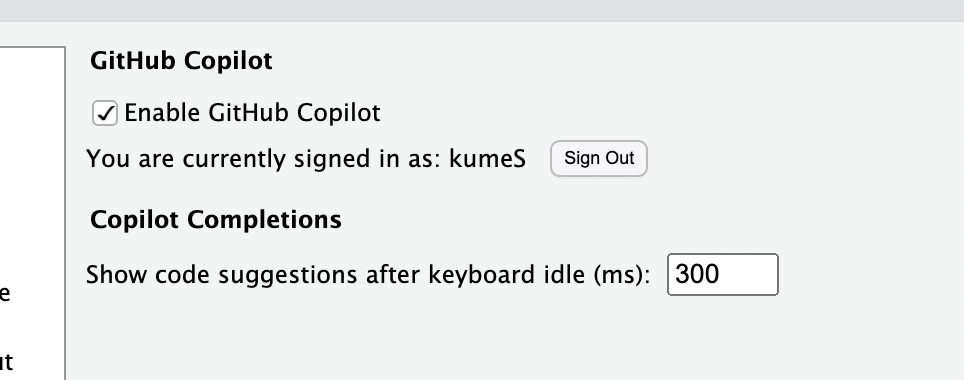
# ヘルプ表示
$ interpreter -h
usage: interpreter [-h][-s SYSTEM_MESSAGE][-l][-y][-d][-m MODEL][-t TEMPERATURE][-c CONTEXT_WINDOW][-x MAX_TOKENS][-b MAX_BUDGET][-ab API_BASE][-ak API_KEY][--config][--conversations][-f]
Open Interpreter
options:
-h, --help show this help message and exit-s SYSTEM_MESSAGE, --system_message SYSTEM_MESSAGE
prompt / custom instructions for the language model
-l, --local run inlocal mode-y, --auto_run automatically run the interpreter
-d, --debug_mode run in debug mode
-m MODEL, --model MODEL
model to use for the language model
-t TEMPERATURE, --temperature TEMPERATURE
optional temperature setting for the language model
-c CONTEXT_WINDOW, --context_window CONTEXT_WINDOW
optional context window size for the language model
-x MAX_TOKENS, --max_tokens MAX_TOKENS
optional maximum number of tokens for the language
model
-b MAX_BUDGET, --max_budget MAX_BUDGET
optionally set the max budget (in USD)for your llm
calls
-ab API_BASE, --api_base API_BASE
optionally set the API base URL for your llm calls(this will override environment variables)-ak API_KEY, --api_key API_KEY
optionally set the API key for your llm calls (this
will override environment variables)--config open config.yaml file in text editor
--conversations list conversations to resume
-f, --fast(depracated) runs `interpreter --model gpt-3.5-turbo`
#litellmのアップグレード(litellm-0.9.2)
/opt/homebrew/bin/pip3.11 install --upgrade litellm
#...
#ERROR: pip's dependency resolver does not currently take into account all the packages that are installed. This behaviour #is the source of the following dependency conflicts.
#open-interpreter 0.1.9 requires litellm<0.8.0,>=0.7.5, but you have litellm 0.9.2 which is incompatible.
#Successfully installed litellm-0.9.2
───────────────────────────────────────────────────────────────────────────────────────────────
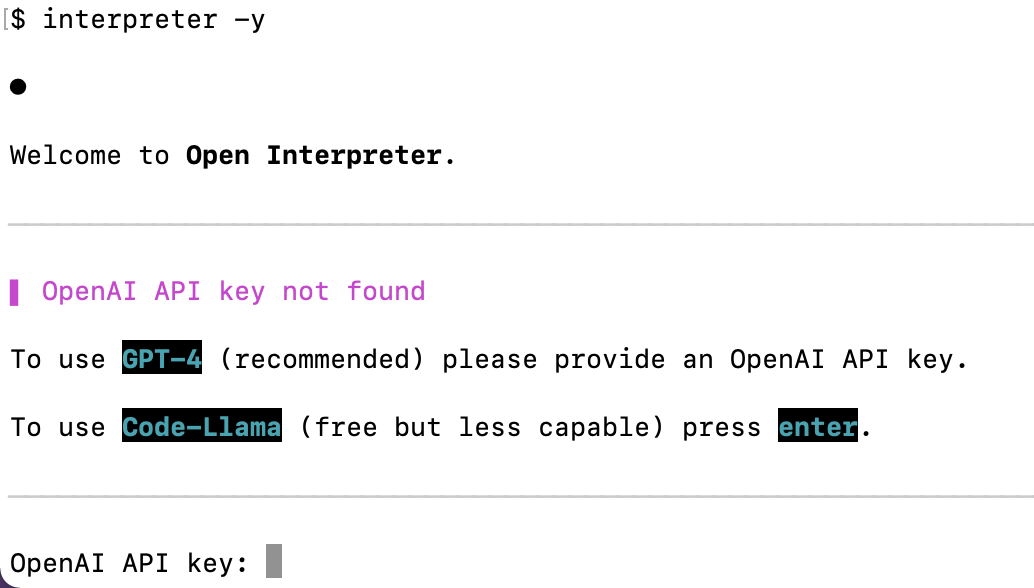
▌ OpenAI API key not found
To use GPT-4 (recommended) please provide an OpenAI API key.
To use Code-Llama (free but less capable) press enter.
───────────────────────────────────────────────────────────────────────────────────────────────
OpenAI API key:
▌ Switching to Code-Llama...
Tip: Run interpreter --local to automatically use Code-Llama.
───────────────────────────────────────────────────────────────────────────────────────────────
Open Interpreter will use Code Llama forlocal execution. Use your arrow keys to set up the
model.
[?] Parameter count (smaller is faster, larger is more capable): 7B
> 7B
13B
34B
[?] Quality (smaller is faster, larger is more capable): Small | Size: 2.6 GB, Estimated RAM us[?] Quality (smaller is faster, larger is more capable): Medium | Size: 3.8 GB, Estimated RAM u[?] Quality (smaller is faster, larger is more capable): Small | Size: 2.6 GB, Estimated RAM us[?] Quality (smaller is faster, larger is more capable): Small | Size: 2.6 GB, Estimated RAM us[?] Quality (smaller is faster, larger is more capable): Medium | Size: 3.8 GB, Estimated RAM u[?] Quality (smaller is faster, larger is more capable): Small | Size: 2.6 GB, Estimated RAM usage: 5.1 GBSize: 2.6 GB, Estimated RAM usage: 5.1 GB
> Small | Size: 2.6 GB, Estimated RAM usage: 5.1 GB
Medium | Size: 3.8 GB, Estimated RAM usage: 6.3 GB
Large | Size: 6.7 GB, Estimated RAM usage: 9.2 GB
See More
[?] Use GPU? (Large models might crash on GPU, but will run more quickly)(Y/n): n
This language model was not found on your system.
Download to `/Users/sas/Library/Application Support/Open Interpreter/models`?
[?](Y/n): n
Download cancelled. Exiting.
```