
- はじめに
- NCBIのE-utilitiesサービスを使った、RefSeqのダウンロード方法について
- 枯草菌(Bacillus subtilis)のRefSeqのダウンロード実行について
- 黄色ブドウ球菌(Staphylococcus aureus)のRefSeqのダウンロード実行
- まとめ
- 補足
- ヒトリファレンスゲノム: GRCh38とhg38の違い
- 参考資料
はじめに
今回、NCBI (National Center for Biotechnology Information, アメリカ国立生物工学情報センター)のリファレンスゲノム、通称「RefSeq」のダウンロード方法についての記事を書いてみました。
NCBIウェブサイトからFTPサイト(ファイル転送プロトコル / File Transfer Protocol((ネットワーク上でファイル等の転送を行う通信プロトコルの1つです。)))を介さずに、RefSeqのFASTA形式ファイルをダウンロードします。
NCBIのRefSeqは、NCBI FTPサイトで公開されており、通常、そのウェブ・インターフェイスから取得します。 ただ、FTPサイトの動作が重かったり、そもそも、FTPサイトに公開されていないRefSeqデータもあります。 そういうときのやり方です。
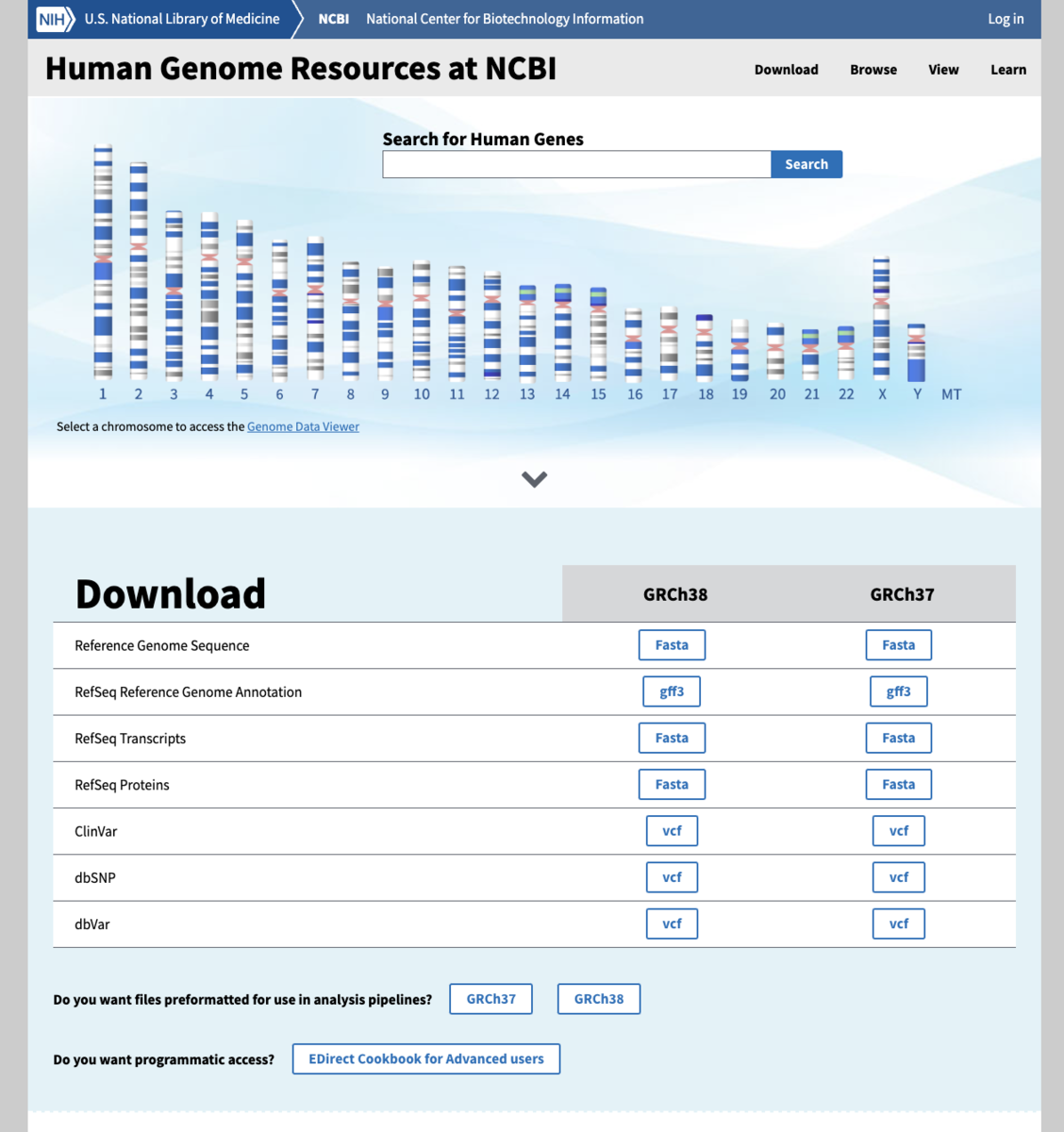
また、Humanゲノムの場合だと、特設のウェブページがあって、GRCh38やGRCh37といった、RefSeqやアノテーションファイルなどが簡単に取得できます。「Human Genome Resources and Download」をクリックすれば、そのページに移動します。
この記事では、NCBIのFTPサイトを介さずに、E-utilitiesサービスを使った代替方法を取り扱います。
NCBI RefSeqのFTPサイトについて
「RefSeq: NCBI Reference Sequence Database」のカテゴリの1つである「RefSeq Access」から、 RefSeq FTPあるいはRefSeq genomes FTPの項目をクリックすることで、FTPサイトを訪れられます。
NCBIのE-utilitiesサービスを使った、RefSeqのダウンロード方法について
NCBIのE-utilitiesというサービスを利用して、RefSeqのテキストデータを取得します。
E-utilities(Entrez Programming Utilities)は、NCBIのEntrezデータベースシステムのAPIのことを言います。 現在、E-utilitiesは、ヌクレオチドおよびタンパク質配列、遺伝子レコード、3次元分子構造、生物医学文献を含む、様々な生物医学データを網羅する38つのデータベースで使われているインターフェイスのようです。
E-utilitiesは、以下のようなURL記法で利用します。
{API名}と{各パラメータ}の部分が変数となっています。
https://eutils.ncbi.nlm.nih.gov/entrez/eutils/{API名}.fcgi?{各パラメータ}
今回、efetch.fcgiのAPIを使用しています。 また、データベースはnuccoreを指定して、fastaファイルを取得するようにしています。
より詳しくは、まさなみブログの「NCBIのE-utilsについて簡単にまとめてみた。」や英語マニュアル「A General Introduction to the E-utilities」などを参照ください。
つづいて、RefSeqのダウンロードについての実施例を紹介します。
枯草菌(Bacillus subtilis)のRefSeqのダウンロード実行について
枯草菌について
枯草菌は、土壌や植物など、比較的どこにでもいる常在細菌の一種です。 この和名は、枯れた草の表面などからよく分離されたことから、その名前が付けられたようです。
Bacillus subtilis subsp. subtilis str. 168 complete genome (NC_000964.3)
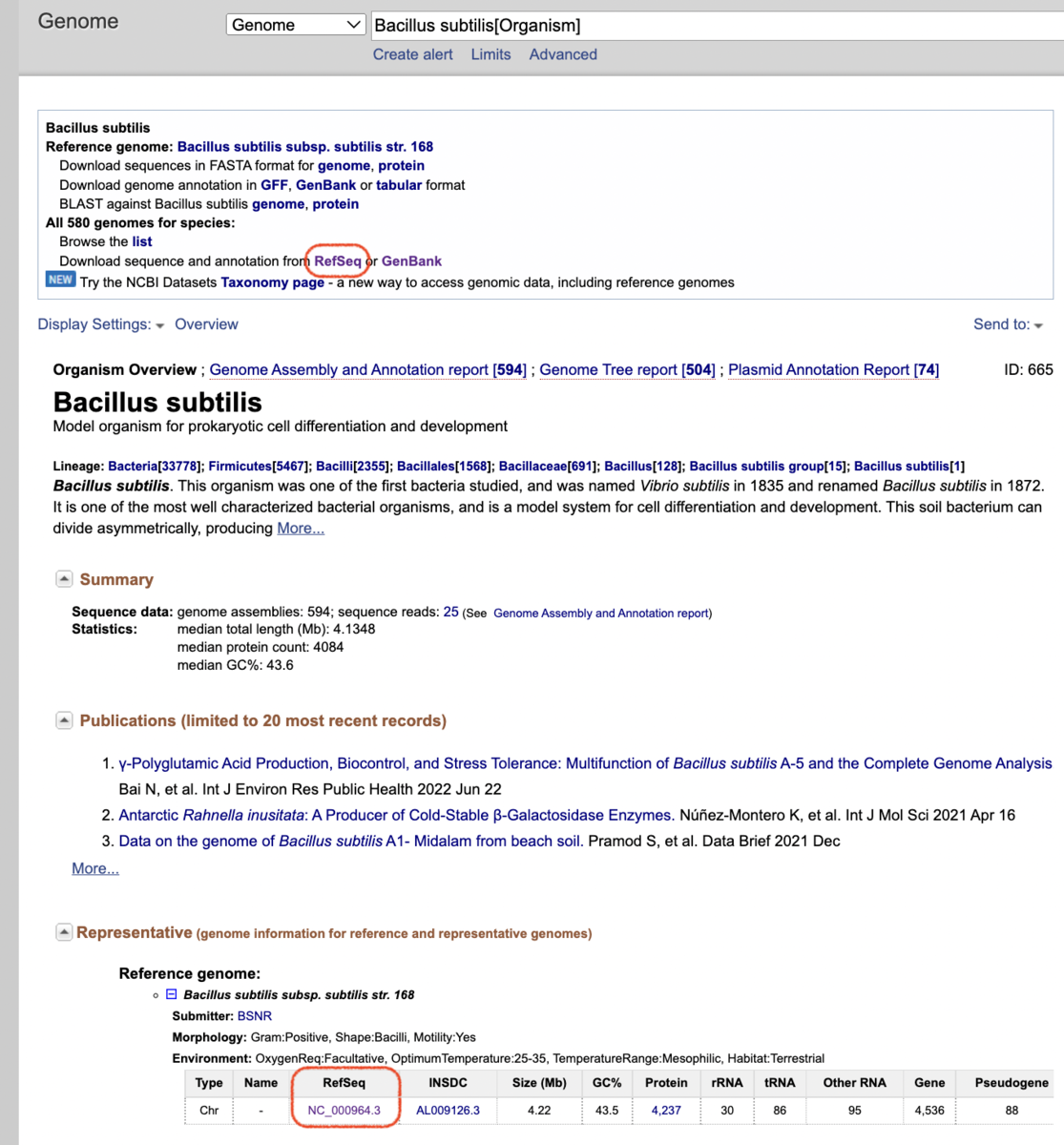
上図の「Bacillus subtilis: Model organism for prokaryotic cell differentiation and development」を対象に、RefSeqのFastaファイルをダウンロードします。NCBI Reference Sequenceは、「NC_000964.3」です。
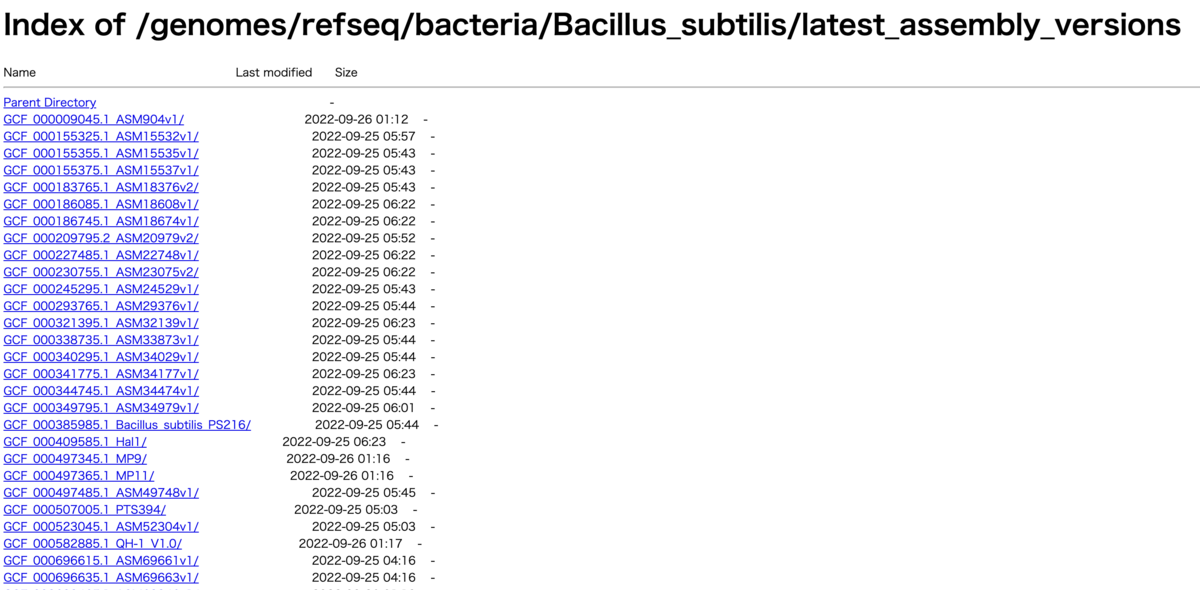
ちなみに、「Download sequence and annotation from RefSeq」の「RefSeq」をクリックすると、 FTPサイトに移動します。Bacillus subtilisのゲノムは沢山表示されます。
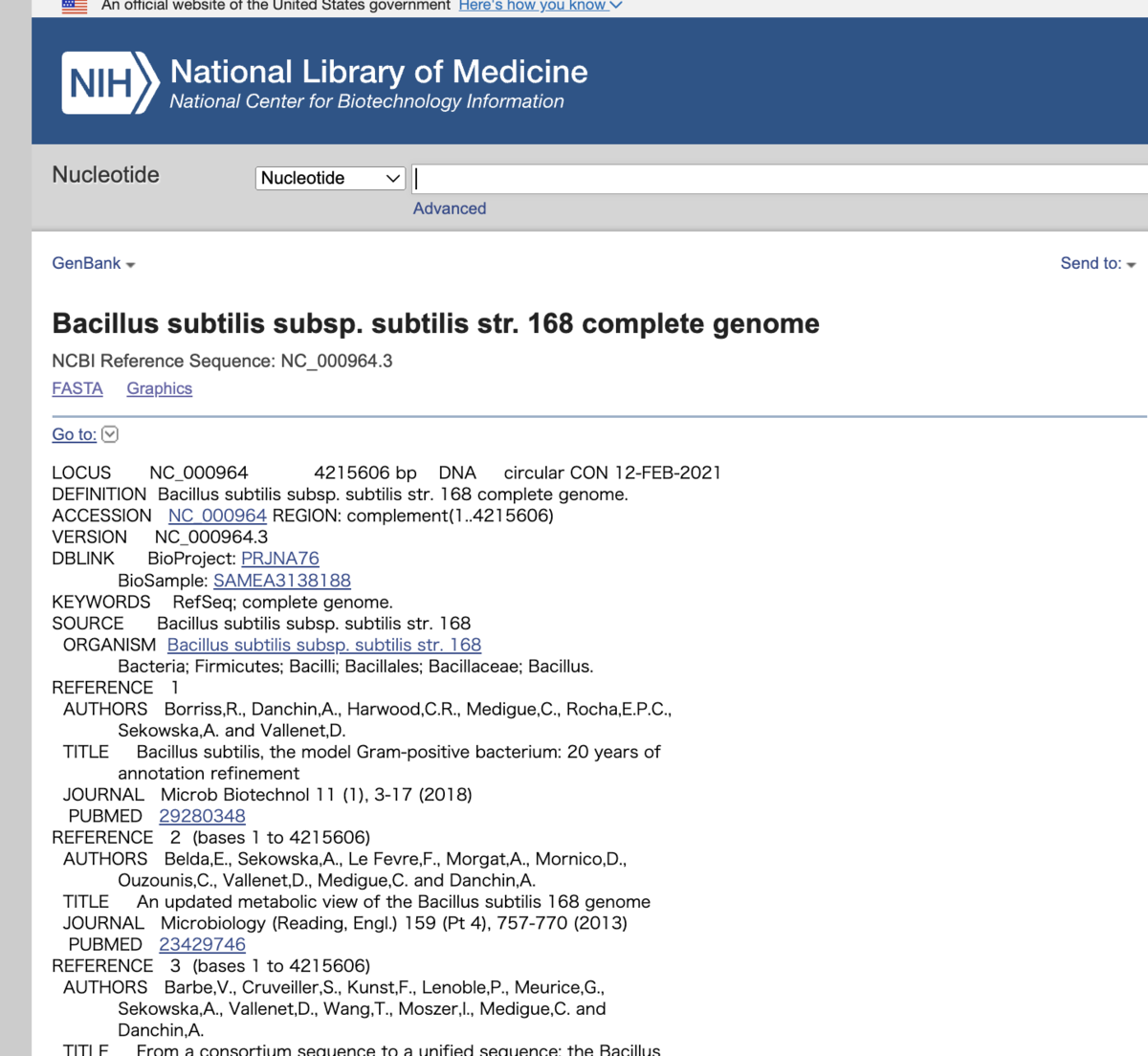
次に、Reference genomeのページに移動します。そして、RefSeq「NC_000964.3」のリンクをクリックします。 そうすると、「Bacillus subtilis subsp. subtilis str. 168 complete genome (NC_000964.3)」が表示されます。 さらに、FASTAのリンクをクリックすると、FASTA形式の塩基配列が表示されます。

この配列データをE-utilitiesサービスを用いてダウンロードします。
Rでの実行コード
まずは、wgetコマンドとR実行を組み合わせて書くと、以下のようになります。
## wgetのR実行の場合 RefSeq_id <- "NC_000964.3" OutFile <- paste0(RefSeq_id, ".fa") #URL作成 url <- paste0('"https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=', RefSeq_id, '&rettype=fasta" -O ', OutFile) #実行 system(paste0("wget -q ", url)) #ファイル情報表示 file.info(OutFile) # size isdir mode mtime ctime #NC_000964.3.fa 4275902 FALSE 644 2022-09-26 19:45:33 2022-09-27 02:32:52 # atime uid gid uname grname #NC_000964.3.fa 2022-09-29 02:18:20 502 20 sas staff
一方、純粋に、R環境のみで実行するなら、utils::download.file関数を使います。 結果は、上記と同じくなりますが。。以下のように実行します。
##utils::download.file()を使った場合 RefSeq_id <- "NC_000964.3" OutFile <- paste0(RefSeq_id, ".fa") #URL作成 url <- paste0("https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=", RefSeq_id, "&rettype=fasta") #実行 utils::download.file(url, OutFile, quiet = TRUE) #ファイル情報表示 file.info(OutFile)
プログラム実行の結果、「NC_000964.3.fa」という名前で、4.3MBのファイルがダウンロードされます。 これくらいのファイルサイズなら、テキストエディタで余裕で開けます。
黄色ブドウ球菌(Staphylococcus aureus)のRefSeqのダウンロード実行
黄色ブドウ球菌について
黄色ブドウ球菌は、ブドウの房状に複数の細菌が集まっていることから、その名前が付けられました。 この細菌は、しばしば、食中毒の原因となります。
もっと詳しい情報は、ようこそ不思議な細菌の世界へ、食中毒を起こす微生物 » 黄色ブドウ球菌とかを参考にしてください。
Staphylococcus aureus subsp. aureus NCTC 8325 chromosome, complete genome (NC_007795.1)

上図の「Staphylococcus aureus subsp. aureus NCTC 8325 chromosome, complete genome」を対象に、RefSeqのFastaファイルをダウンロードします。NCBI Reference Sequenceは、「NC_007795.1」です。

ちなみに、「Staphylococcus aureus」のFTPサイトでは何もRefSeqが表示されません。
Rでの実行コード
このFATSAファイルのダウンロードでは、上記のRコードを関数化したものを使います。 関数の定義と実行を以下に示します。引数は、NCBI Reference SequenceのIDを入力するだけです。
## efetch.fcgiの実行関数 download.RefSeq() download.RefSeq <- function(NCBI_Reference_Sequence){ #定義 RefSeq_id <- NCBI_Reference_Sequence OutFile <- paste0(RefSeq_id, ".fa") #URL作成 url <- paste0("https://eutils.ncbi.nlm.nih.gov/entrez/eutils/efetch.fcgi?db=nuccore&id=", RefSeq_id, "&rettype=fasta") #実行 utils::download.file(url, OutFile, quiet = TRUE) #ファイル情報表示 file.info(OutFile) } #関数実行 download.RefSeq(NCBI_Reference_Sequence="NC_007795.1") # size isdir mode mtime ctime #NC_007795.1.fa 2861755 FALSE 644 2022-09-27 02:41:14 2022-09-27 02:41:14 # atime uid gid uname grname #NC_007795.1.fa 2022-09-27 02:40:47 502 20 sas staff

プログラム実行の結果、「NC_007795.1.fa」という名前で、 2.9MBのファイルがダウンロードされます。
まとめ
E-utilitiesとか、R言語を使うと、スマートに出来ますよね。
場合によっては、RefSeqのテキストをコピーして、 テキストエディタに貼り付けて保存すると同じことなんだけど。。。
補足
wgetのインストール
Macにwgetをインストールしていない場合には、まずは、wgetをインストールします。
ターミナル環境でインストールする場合には、
#(ターミナル)brewコマンドでのwgetのインストール
brew install wget
また、R環境でインストールする場合には、
#(R環境)brewコマンドでのwgetのインストール system("brew install wget")
ヒトリファレンスゲノム: GRCh38とhg38の違い
GRCh38は、NCBI/GRCバージョンを意味します。hg38は、UCSCバージョンを意味します。
GRCh38とhg38の配列情報はほぼ同じです。曖昧な塩基表記に違いがあります。hg38はNで表記される一方、GRCh38はあいまい塩基表記(R,Y等)を記述されます。
両者で、ミトコンドリアの配列情報は同じで、座標のずれは全染色体通してありません。
参考資料
https://cell-innovation.nig.ac.jp/SurfWiki/GRCh38_Hg38_defference.html