
- 概要
- イントロダクション
- Bioconductorパッケージのインストール
- データ・ダウンロード
- セットアップ
- 遺伝子のフィルタリング
- 正規化
- 探索的データ解析
- 線形モデルの構築
- 発現変動を調べる
- 結果を見る
- 変動遺伝子を可視化する
- 遺伝子セットのエンリッチメント評価
- 生命科学データ・RNA-seq解析についての関連図書
- 参考資料
概要
R言語を使った、RNA-seq解析チュートリアルを作成してみた。
データは、RNA-seqのカウント済みのデータを使用している。
本記事は、以下のArticleの日本語訳記事 + α である。
# HGEN 473 - Genomics # Spring 2017 # Tuesday, May 9 & Thursday, May 11 # RNA-seq analysis with R/Bioconductor # John Blischak # Last updated: 2020-04-08 # https://gist.github.com/jdblischak/fdb1745612927252a7633751e5e60bcb
イントロダクション
このチュートリアルは、Bioconductorが提供するオープンソース・パッケージを使った、RNA-seqデータ解析を紹介する。
今回使用するRコードは、Charity Law et al.の論文「RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR」(2017年)から引用している。原書論文、及びその派生作品は、CC-BY4.0ライセンスで自由に利用できる。
# 原著: https://f1000research.com/articles/5-1408/v2 # ソースコード: https://bioconductor.org/packages/release/workflows/html/RNAseq123.html # 引用: Law CW, Alhamdoosh M, Su S et al. RNA-seq analysis is easy as 1-2-3 # with limma, Glimma and edgeR [version 2; referees: 3 approved]. # F1000Research 2016, 5:1408 (doi: 10.12688/f1000research.9005.2)
ゲノム解析の関連記事
Bioconductor - RNAseq123
Bioconductorパッケージのインストール
このチュートリアルで使用するBioconductorやCRANのパッケージ群をインストールする。
以下のインストールコマンドを実行して、「RNAseq123」ワークフローパッケージをインストールします。
install.packages("BiocManager") BiocManager::install("RNAseq123") #Update all/some/none? [a/s/n]: #と聞かれたら a を入力して、Enter #Do you want to install from sources the packages which need compilation? (Yes/no/cancel) #と聞かれたら Yes を入力して、Enter install.packages("Matrix") #Do you want to install from sources the package which needs compilation? (Yes/no/cancel) no #>> #と聞かれたら no を入力して、Enter #ワークフローパッケージ「RNAseq123」を読み込む library(RNAseq123)
データ・ダウンロード
このサンプルデータは、Sheridanら(BMC Cancer, 2015)が収集した、マウス乳腺の3つの細胞集団におけるRNA-seq結果である。
以下のコードでは、Gene Expression Omnibus(GEO, NCBIのゲノミクスデータ・リポジトリ)からカウントデータをダウンロードしている。
#URL定義 url <- "https://www.ncbi.nlm.nih.gov/geo/download/?acc=GSE63310&format=file" #ダウンロード utils::download.file(url, destfile = "GSE63310_RAW.tar", mode = "wb") #OR utils::download.file(url, destfile = "GSE63310_RAW.tar", mode = "wb", method = "wget") #OR utils::download.file(url, destfile = "GSE63310_RAW.tar", mode = "wb", method = "curl") #tarファイル解凍 utils::untar("GSE63310_RAW.tar", exdir = ".") #gzファイル解凍 files_gz <- Sys.glob("GSM*txt.gz") files_gz # [1] "GSM1545535_10_6_5_11.txt.gz" # [2] "GSM1545536_9_6_5_11.txt.gz" # [3] "GSM1545537_mo906111-1_m09611-2.txt.gz" # [4] "GSM1545538_purep53.txt.gz" # [5] "GSM1545539_JMS8-2.txt.gz" # [6] "GSM1545540_JMS8-3.txt.gz" # [7] "GSM1545541_JMS8-4.txt.gz" # [8] "GSM1545542_JMS8-5.txt.gz" # [9] "GSM1545543_JMS9-CDBG.txt.gz" #[10] "GSM1545544_JMS9-P7c.txt.gz" #[11] "GSM1545545_JMS9-P8c.txt.gz" for(f in files_gz){ R.utils::gunzip(f, overwrite = TRUE) } #ファイル確認 dir(pattern = ".txt") # [1] "GSM1545535_10_6_5_11.txt" "GSM1545536_9_6_5_11.txt" # [3] "GSM1545537_mo906111-1_m09611-2.txt" "GSM1545538_purep53.txt" # [5] "GSM1545539_JMS8-2.txt" "GSM1545540_JMS8-3.txt" # [7] "GSM1545541_JMS8-4.txt" "GSM1545542_JMS8-5.txt" # [9] "GSM1545543_JMS9-CDBG.txt" "GSM1545544_JMS9-P7c.txt" #[11] "GSM1545545_JMS9-P8c.txt"
データセットの引用元を下記に示す。
# データセットの引用: # Sheridan JM, Ritchie ME, Best SA, et al.: A pooled shRNA screen for # regulators of primary mammary stem and progenitor cells identifies # roles for Asap1 and Prox1. BMC Cancer. 2015; 15(1): 221.
セットアップ
このセクションでは、TXTファイルのカウントデータを読み込んで、各種のアノテーションを付与する。
関連パッケージの読み込み
#線形モデルによる発現量変動解析 library("limma") #探索のためのインタラクティブなプロット library("Glimma") #NGS実験のカウントデータの処理 library("edgeR") #Mus musculus ゲノムへの遺伝子アノテーション library("Mus.musculus")
TXTデータの読み込み
#ファイル名の定義 files <- c("GSM1545535_10_6_5_11.txt", "GSM1545536_9_6_5_11.txt", "GSM1545538_purep53.txt", "GSM1545539_JMS8-2.txt", "GSM1545540_JMS8-3.txt", "GSM1545541_JMS8-4.txt", "GSM1545542_JMS8-5.txt", "GSM1545544_JMS9-P7c.txt", "GSM1545545_JMS9-P8c.txt") #試しに、先頭5行の読み込み read.delim(files[1], nrow = 5) # EntrezID GeneLength Count #1 497097 3634 1 #2 100503874 3259 0 #3 100038431 1634 0 #4 19888 9747 0 #5 20671 3130 1
今回、EntrezIDとCountの列(1列目と3列目)を使う。
#edgeR::readDGEで、カウントデータを含むファイル群を読み込んで、データ結合させる。 x <- edgeR::readDGE(files, columns = c(1, 3)) class(x) #[1] "DGEList" #attr(,"package") #[1] "edgeR" dim(x) #[1] 27179 9 names(x) #[1] "samples" "counts" str(x) #Formal class 'DGEList' [package "edgeR"] with 1 slot # ..@ .Data:List of 2 # .. ..$ :'data.frame': 9 obs. of 4 variables: # .. .. ..$ files : chr [1:9] "GSM1545535_10_6_5_11.txt" "GSM1545536_9_6_5_11.txt" "GSM1545538_purep53.txt" "GSM1545539_JMS8-2.txt" ... # .. .. ..$ group : Factor w/ 1 level "1": 1 1 1 1 1 1 1 1 1 # .. .. ..$ lib.size : num [1:9] 32863052 35335491 57160817 51368625 75795034 ... # .. .. ..$ norm.factors: num [1:9] 1 1 1 1 1 1 1 1 1 # .. ..$ : num [1:27179, 1:9] 1 0 0 0 1 431 768 4 810 452 ... # .. .. ..- attr(*, "dimnames")=List of 2 # .. .. .. ..$ Tags : chr [1:27179] "497097" "100503874" "100038431" "19888" ... # .. .. .. ..$ Samples: chr [1:9] "GSM1545535_10_6_5_11" "GSM1545536_9_6_5_11" "GSM1545538_purep53" "GSM1545539_JMS8-2" ...
サンプルにアノテーションをつける。
x$samples # files group lib.size norm.factors #GSM1545535_10_6_5_11 GSM1545535_10_6_5_11.txt 1 32863052 1 #GSM1545536_9_6_5_11 GSM1545536_9_6_5_11.txt 1 35335491 1 #GSM1545538_purep53 GSM1545538_purep53.txt 1 57160817 1 #GSM1545539_JMS8-2 GSM1545539_JMS8-2.txt 1 51368625 1 #GSM1545540_JMS8-3 GSM1545540_JMS8-3.txt 1 75795034 1 #GSM1545541_JMS8-4 GSM1545541_JMS8-4.txt 1 60517657 1 #GSM1545542_JMS8-5 GSM1545542_JMS8-5.txt 1 55086324 1 #GSM1545544_JMS9-P7c GSM1545544_JMS9-P7c.txt 1 21311068 1 #GSM1545545_JMS9-P8c GSM1545545_JMS9-P8c.txt 1 19958838 1 colnames(x) #[1] "GSM1545535_10_6_5_11" "GSM1545536_9_6_5_11" #[3] "GSM1545538_purep53" "GSM1545539_JMS8-2" #[5] "GSM1545540_JMS8-3" "GSM1545541_JMS8-4" #[7] "GSM1545542_JMS8-5" "GSM1545544_JMS9-P7c" #[9] "GSM1545545_JMS9-P8c" #「_」の後ろにくる文字列を抽出する samplenames <- substring(colnames(x), 12, nchar(colnames(x))) samplenames #[1] "10_6_5_11" "9_6_5_11" "purep53" "JMS8-2" "JMS8-3" #[6] "JMS8-4" "JMS8-5" "JMS9-P7c" "JMS9-P8c" #列名に付け替える colnames(x) <- samplenames #グループ列を書き換える x$samples$group #[1] 1 1 1 1 1 1 1 1 1 #Levels: 1 group <- as.factor(c("LP", "ML", "Basal", "Basal", "ML", "LP", "Basal", "ML", "LP")) x$samples$group <- group #Laneを追加する lane <- as.factor(rep(c("L004", "L006", "L008"), c(3, 4, 2))) x$samples$lane <- lane #x$samplesの表示 x$samples # files group lib.size norm.factors lane #10_6_5_11 GSM1545535_10_6_5_11.txt LP 32863052 1 L004 #9_6_5_11 GSM1545536_9_6_5_11.txt ML 35335491 1 L004 #purep53 GSM1545538_purep53.txt Basal 57160817 1 L004 #JMS8-2 GSM1545539_JMS8-2.txt Basal 51368625 1 L006 #JMS8-3 GSM1545540_JMS8-3.txt ML 75795034 1 L006 #JMS8-4 GSM1545541_JMS8-4.txt LP 60517657 1 L006 #JMS8-5 GSM1545542_JMS8-5.txt Basal 55086324 1 L006 #JMS9-P7c GSM1545544_JMS9-P7c.txt ML 21311068 1 L008 #JMS9-P8c GSM1545545_JMS9-P8c.txt LP 19958838 1 L008
ここで、lib.sizeは、ライブラリーサイズ; トータルのリード数である。
norm.factorsは、正規化係数である。
遺伝子をアノテーションする
head(x$counts) # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 1 2 342 526 3 3 535 2 0 # 100503874 0 0 5 6 0 0 5 0 0 # 100038431 0 0 0 0 0 0 1 0 0 # 19888 0 1 0 0 17 2 0 1 0 # 20671 1 1 76 40 33 14 98 18 8 # 27395 431 771 1368 1268 1564 769 818 468 342 dim(x$counts) #[1] 27179 9 #Tags情報(Entrez Gene ID)を取得する geneid <- rownames(x) head(geneid) #[1] "497097" "100503874" "100038431" "19888" "20671" "27395"
次に、マウスのアノテーション情報を使用して、Gene IDとGene SYMBOL、TXCHROMとを関係づける。
Mus.musculus #OrganismDb Object: # Includes GODb Object: GO.db # With data about: Gene Ontology # Includes OrgDb Object: org.Mm.eg.db # Gene data about: Mus musculus # Taxonomy Id: 10090 # Includes TxDb Object: TxDb.Mmusculus.UCSC.mm10.knownGene # Transcriptome data about: Mus musculus # Based on genome: mm10 # The OrgDb gene id ENTREZID is mapped to the TxDb gene id GENEID . genes <- AnnotationDbi::select(Mus.musculus, keys = geneid, columns = c("SYMBOL", "TXCHROM"), keytype = "ENTREZID") head(genes) # ENTREZID SYMBOL TXCHROM #1 497097 Xkr4 chr1 #2 100503874 Gm19938 <NA> #3 100038431 Gm10568 <NA> #4 19888 Rp1 chr1 #5 20671 Sox17 chr1 #6 27395 Mrpl15 chr1 #Gene IDが重複している箇所を除く genes <- genes[!duplicated(genes$ENTREZID), ] #Gene ID情報を付与する x$genes <- genes x #An object of class "DGEList" #$samples # files group lib.size norm.factors lane #10_6_5_11 GSM1545535_10_6_5_11.txt LP 32863052 1 L004 #9_6_5_11 GSM1545536_9_6_5_11.txt ML 35335491 1 L004 #purep53 GSM1545538_purep53.txt Basal 57160817 1 L004 #JMS8-2 GSM1545539_JMS8-2.txt Basal 51368625 1 L006 #JMS8-3 GSM1545540_JMS8-3.txt ML 75795034 1 L006 #JMS8-4 GSM1545541_JMS8-4.txt LP 60517657 1 L006 #JMS8-5 GSM1545542_JMS8-5.txt Basal 55086324 1 L006 #JMS9-P7c GSM1545544_JMS9-P7c.txt ML 21311068 1 L008 #JMS9-P8c GSM1545545_JMS9-P8c.txt LP 19958838 1 L008 # #$counts # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 1 2 342 526 3 3 535 2 0 # 100503874 0 0 5 6 0 0 5 0 0 # 100038431 0 0 0 0 0 0 1 0 0 # 19888 0 1 0 0 17 2 0 1 0 # 20671 1 1 76 40 33 14 98 18 8 #27174 more rows ... # #$genes # ENTREZID SYMBOL TXCHROM #1 497097 Xkr4 chr1 #2 100503874 Gm19938 <NA> #3 100038431 Gm10568 <NA> #4 19888 Rp1 chr1 #5 20671 Sox17 chr1 #27174 more rows ...
遺伝子のフィルタリング
counts-per-million (CPM) を算出(log)する
cpmは、解析対象のサンプルすべてに対して、総リード数を 100 万に揃える正規化法である。
#CPMの実行 cpm <- edgeR::cpm(x) head(cpm) # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 0.03042931 0.05660032 5.98311952 10.2397134 0.03958043 0.04957231 9.71202943 0.09384795 0.0000000 # 100503874 0.00000000 0.00000000 0.08747251 0.1168028 0.00000000 0.00000000 0.09076663 0.00000000 0.0000000 # 100038431 0.00000000 0.00000000 0.00000000 0.0000000 0.00000000 0.00000000 0.01815333 0.00000000 0.0000000 # 19888 0.00000000 0.02830016 0.00000000 0.0000000 0.22428910 0.03304821 0.00000000 0.04692397 0.0000000 # 20671 0.03042931 0.02830016 1.32958212 0.7786854 0.43538472 0.23133744 1.77902595 0.84463153 0.4008249 # 27395 13.11503265 21.81942229 23.93247808 24.6843282 20.63459725 12.70703524 14.84942070 21.96041982 17.1352661 lcpm <- edgeR::cpm(x, log = TRUE) head(lcpm) # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 -3.748623 -3.313765 2.5914607 3.362285 -3.581259 -3.418282 3.2862891 -2.8591947 -4.507432 # 100503874 -4.507432 -4.507432 -2.9275280 -2.636931 -4.507432 -4.507432 -2.8918170 -4.5074315 -4.507432 # 100038431 -4.507432 -4.507432 -4.5074315 -4.507432 -4.507432 -4.507432 -4.0087883 -4.5074315 -4.507432 # 19888 -4.507432 -3.790514 -4.5074315 -4.507432 -1.898317 -3.698711 -4.5074315 -3.4597175 -4.507432 # 20671 -3.748623 -3.790514 0.4579085 -0.281645 -1.060843 -1.860900 0.8663089 -0.1703963 -1.168797 # 27395 3.717978 4.450445 4.5835457 4.628091 4.370064 3.672539 3.8965999 4.4597191 4.102594 # 9サンプルすべてで、ゼロカウントの遺伝子を検出する head(x$counts == 0) # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE TRUE # 100503874 TRUE TRUE FALSE FALSE TRUE TRUE FALSE TRUE TRUE # 100038431 TRUE TRUE TRUE TRUE TRUE TRUE FALSE TRUE TRUE # 19888 TRUE FALSE TRUE TRUE FALSE FALSE TRUE FALSE TRUE # 20671 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE # 27395 FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE FALSE #5153遺伝子見つかった table(base::rowSums(x$counts == 0) == 9) #FALSE TRUE #22026 5153
次に、遺伝子発現量分布を可視化してみる。
# 遺伝子発現量分布の可視化 limma::plotDensities(lcpm, legend = FALSE, main = "Before filtering") abline(v = 0, lty = 3) #quartz.save(file="Fig01.png", dpi=100, type="png", width=6, height=5)
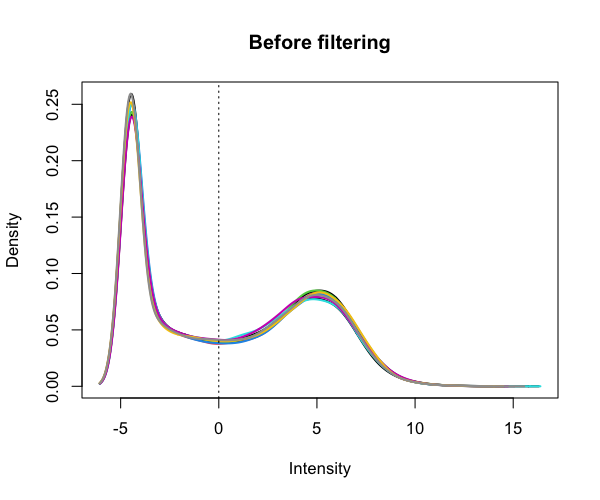
発現量が少ない遺伝子は除外して、処理する。
#少なくとも3つのサンプルで、CPMが1以上である遺伝子のみを残す keep.exprs <- base::rowSums(cpm > 1) >= 3 x1 <- x[keep.exprs, , keep.lib.sizes = FALSE] dim(x1) #[1] 14165 9 #改めて、 log CPM に変換 lcpm <- cpm(x1, log=TRUE) # フィルタリング後の遺伝子発現量分布の可視化 plotDensities(lcpm, legend = FALSE, main = "After filtering") abline(v = 0, lty = 3) #quartz.save(file="Fig02.png", dpi=100, type="png", width=6, height=5)
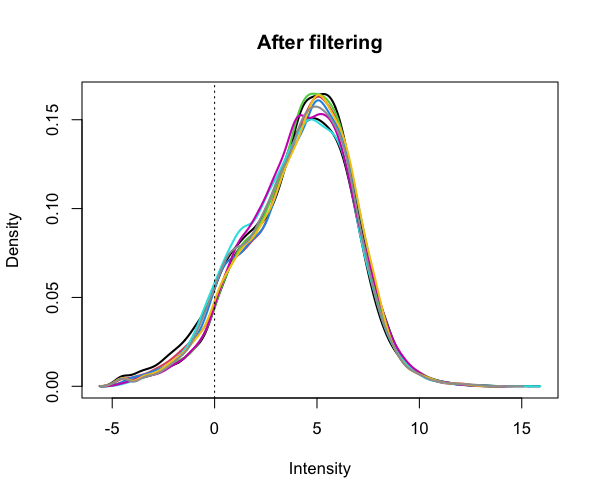
ディスカッション・メモ
Count-per-million (CPM) を使うと、どのような情報が無視されるか?
それは、発現変動分析を行うための有効な測定値であるか?
正規化
edgeRが提供するデフォルトの正規化法は、TMM(trimmed mean of M-values)である。
TMM法では、高発現遺伝子の差異が遺伝子発現分布全体に偏ることを防ぐことができる。
ここで見られるように、しばしば適度な効果をもたらす。
#ライブラリサイズの正規化 x1 <- edgeR::calcNormFactors(x1, method = "TMM") x1$samples$norm.factors #[1] 0.8957309 1.0349196 1.0439552 1.0405040 1.0323599 0.9223424 0.9836603 1.0827381 0.9792607 #ここでは、必要に応じてTMM法が動作することを示すために、 #やや極端な事例を紹介する。 #x2に代入して、擬似的にデータを作成する x2 <- x1 #norm.factorsを 1 にする x2$samples$norm.factors <- 1 #1つ目と2つ目のデータに細工する x2$counts[,1] <- ceiling(x2$counts[, 1] * 0.05) x2$counts[,2] <- x2$counts[, 2] * 5 #そのまま、CPM lcpm2 <- cpm(x2, log = TRUE) boxplot(lcpm2, las = 2, main = "Before normalization") #quartz.save(file="Fig03.png", dpi=100, type="png", width=6, height=5)

#TMMで、正規化 x2 <- calcNormFactors(x2, method = "TMM") x2$samples$norm.factors lcpm2 <- cpm(x2, log = TRUE) boxplot(lcpm2, las=2, main = "After normalization") #quartz.save(file="Fig04.png", dpi=100, type="png", width=6, height=5)

探索的データ解析
多次元尺度法(MDS)でサンプルの関係性を可視化する
library("RColorBrewer") #グループに対応する色パレットを作成する group #[1] LP ML Basal Basal ML LP Basal ML LP #Levels: Basal LP ML col.group <- group levels(col.group) <- brewer.pal(nlevels(col.group), "Set1") col.group <- as.character(col.group) col.group #[1] "#377EB8" "#4DAF4A" "#E41A1C" "#E41A1C" "#4DAF4A" "#377EB8" "#E41A1C" "#4DAF4A" "#377EB8" #レーンに対応する色パレットを作成する lane #[1] L004 L004 L004 L006 L006 L006 L006 L008 L008 #Levels: L004 L006 L008 col.lane <- lane levels(col.lane) <- brewer.pal(nlevels(col.lane), "Set2") col.lane <- as.character(col.lane) col.lane #[1] "#66C2A5" "#66C2A5" "#66C2A5" "#FC8D62" "#FC8D62" "#FC8D62" "#FC8D62" "#8DA0CB" "#8DA0CB"
次に、MDSで関係性を可視化してみる。
#グループでラベリング limma::plotMDS(lcpm, labels = group, col = col.group, main = "group") #quartz.save(file="Fig05.png", dpi=100, type="png", width=6, height=5)
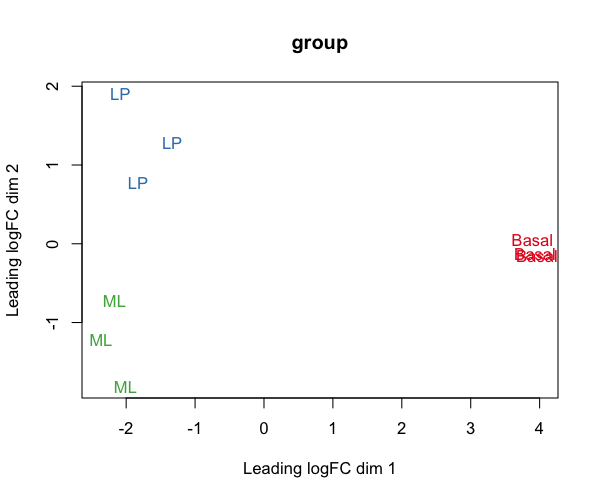
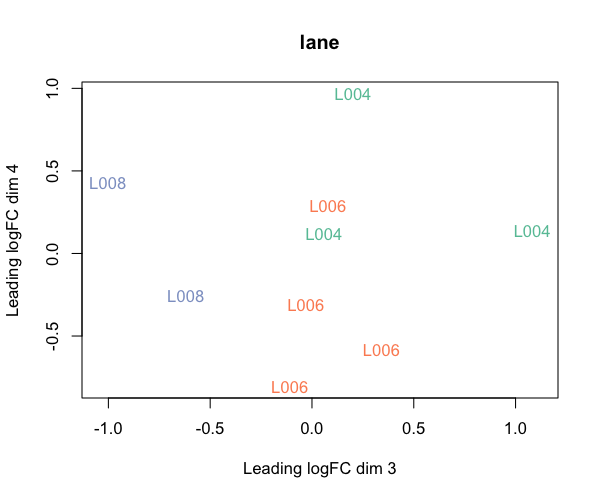
#レーンでラベリング limma::plotMDS(lcpm, labels = lane, col = col.lane, dim = c(3, 4), main = "lane") #quartz.save(file="Fig06.png", dpi=100, type="png", width=6, height=5)
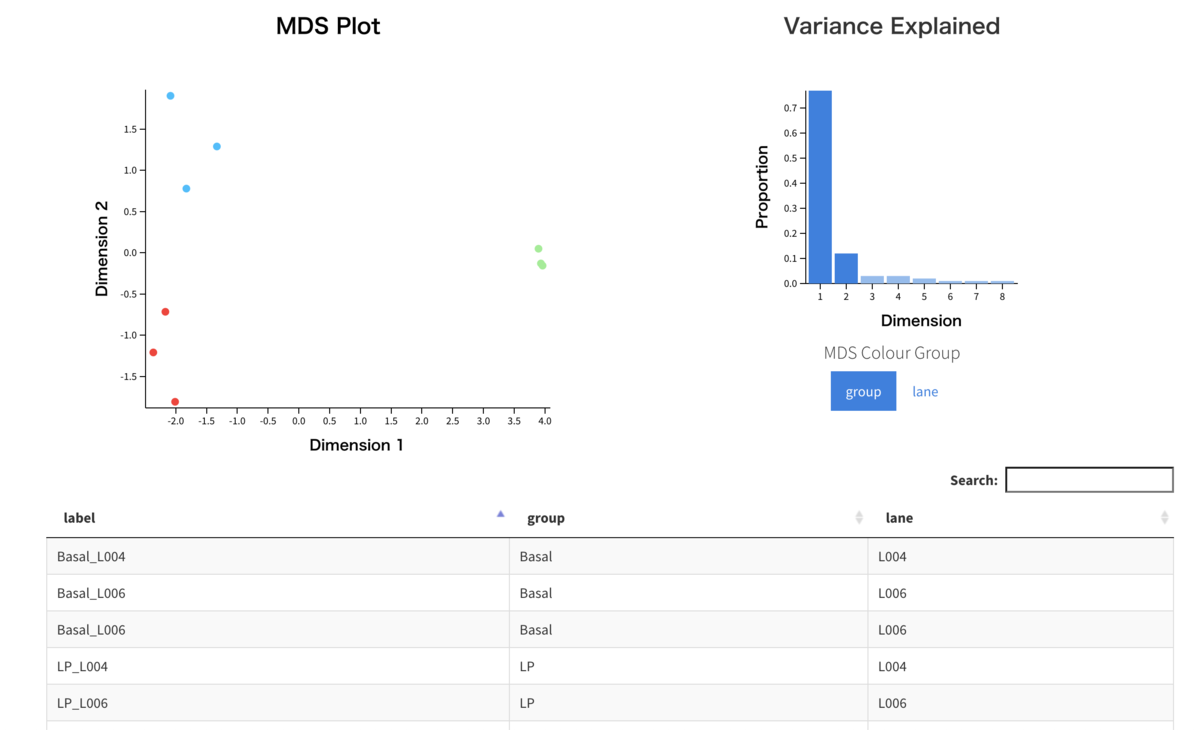
MDSのInteractiveプロット
Glimma::glMDSPlot(lcpm, labels = paste(group, lane, sep = "_"), groups = x1$samples[, c(2, 5)], launch = TRUE) #Fig07.png
線形モデルの構築
今回の線形モデルには、切片(intercept)がない。 これは、limmaユーザーズガイドでは、group-means parametrizationと呼ばれている。 そのグループの平均発現量を、各係数(β)でモデル化することの利点は、 特定の仮説を容易に検証できることである。
design <- stats::model.matrix(~0 + group + lane) colnames(design) <- gsub("group", "", colnames(design)) design # Basal LP ML laneL006 laneL008 #1 0 1 0 0 0 #2 0 0 1 0 0 #3 1 0 0 0 0 #4 1 0 0 1 0 #5 0 0 1 1 0 #6 0 1 0 1 0 #7 1 0 0 1 0 #8 0 0 1 0 1 #9 0 1 0 0 1 #attr(,"assign") #[1] 1 1 1 2 2 #attr(,"contrasts") #attr(,"contrasts")$group #[1] "contr.treatment" #attr(,"contrasts")$lane #[1] "contr.treatment"
下記の図に、モデルを示した。
Yは、特定のサンプルにおける特定遺伝子の発現レベルに対応する。
plot(0, 1, type="n", axes=FALSE, xlab="", ylab="", family = "sans") text(0, 1, labels=expression(Y == italic("\u03b2")[basal]+italic("\u03b2")[LP]+italic("\u03b2")[ML]+italic("\u03b2")[L06]+italic("\u03b2")[L08]+italic("\u03b5")), cex=1.5) #quartz.save(file="Fig08.png", dpi=100, type="png", width=6, height=2.5)

http://www.latex-cmd.com/special/greek.htmlwww.latex-cmd.com
3つの対比を作り、3つの仮説を検証する。
BasalvsLP: Basal細胞とLP細胞の間で発現変動(DE)となる遺伝子は?
BasalvsML: Basal細胞とML細胞の間でDEとなる遺伝子は?
LPvsML: LP細胞とML細胞の間でDEとなる遺伝子は?
#一連のパラメータの特定コントラストに対応するコントラスト行列を構築する contr.matrix <- limma::makeContrasts(BasalvsLP = Basal - LP, BasalvsML = Basal - ML, LPvsML = LP - ML, levels = colnames(design)) contr.matrix # Contrasts #Levels BasalvsLP BasalvsML LPvsML # Basal 1 1 0 # LP -1 0 1 # ML 0 -1 -1 # laneL006 0 0 0 # laneL008 0 0 0
線形モデルで使用されるカウント変換
RNA-seqカウントは、線形モデルに直接使用することはできない。 なぜなら、その前提条件(特に、分散が平均に依存しないこと)に反しているからである。
1つのオプションは、カウントを直接的にモデル化して実行することである(例えば、そのようなモデルは、edgeRやDESeq2で提供されている)。しかし、線形モデリングフレームワークは、一般によりフレキシブルである。limmaパッケージには、データ検証を行う、ナイスなダウンストリーム関数が多数用意されている(例えば、機能カテゴリのエンリッチメント評価など)。
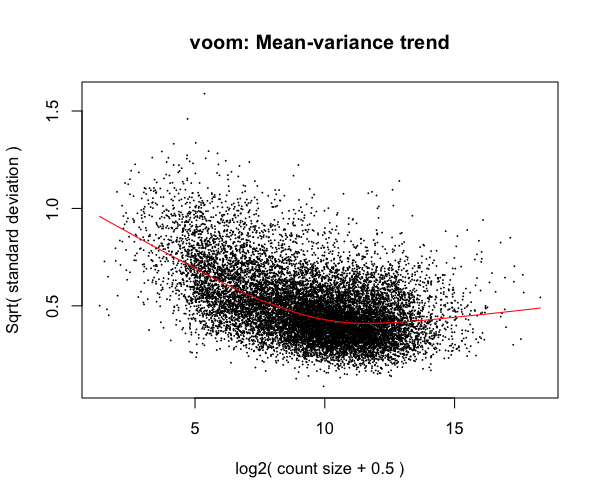
log-CPMへの変換後でさえも、RNA-seqデータには、平均と分散の関係性が残っている。 そこで,voom関数は,この関係性を相殺する重みを計算する。
v <- voom(x1, design, plot = TRUE) #quartz.save(file="Fig09.png", dpi=100, type="png", width=6, height=5)
v #An object of class "EList" #$genes # ENTREZID SYMBOL TXCHROM #1 497097 Xkr4 chr1 #6 27395 Mrpl15 chr1 #7 18777 Lypla1 chr1 #9 21399 Tcea1 chr1 #10 58175 Rgs20 chr1 #14160 more rows ... # #$targets # files group lib.size norm.factors lane #10_6_5_11 GSM1545535_10_6_5_11.txt LP 29409426 0.8957309 L004 #9_6_5_11 GSM1545536_9_6_5_11.txt ML 36528591 1.0349196 L004 #purep53 GSM1545538_purep53.txt Basal 59598629 1.0439552 L004 #JMS8-2 GSM1545539_JMS8-2.txt Basal 53382070 1.0405040 L006 #JMS8-3 GSM1545540_JMS8-3.txt ML 78175314 1.0323599 L006 #JMS8-4 GSM1545541_JMS8-4.txt LP 55762781 0.9223424 L006 #JMS8-5 GSM1545542_JMS8-5.txt Basal 54115150 0.9836603 L006 #JMS9-P7c GSM1545544_JMS9-P7c.txt ML 23043111 1.0827381 L008 #JMS9-P8c GSM1545545_JMS9-P8c.txt LP 19525423 0.9792607 L008 # #$E # Samples #Tags 10_6_5_11 9_6_5_11 purep53 JMS8-2 JMS8-3 JMS8-4 JMS8-5 JMS9-P7c JMS9-P8c # 497097 -4.293244 -3.869026 2.522753 3.302006 -4.481286 -3.993876 3.306782 -3.204336 -5.287282 # 27395 3.875010 4.400568 4.521172 4.570624 4.322845 3.786547 3.918878 4.345642 4.132678 # 18777 4.707695 5.559334 5.400569 5.171235 5.627798 5.081794 5.080061 5.757404 5.150470 # 21399 4.784462 4.741999 5.374548 5.130925 4.848030 4.944024 5.158292 5.036933 4.987679 # 58175 3.943567 3.294875 -1.767924 -1.880302 2.993289 3.357379 -2.114104 3.142621 3.523290 #14160 more rows ... #(中略)
便宜上、voom関数では、log-CPMを計算して、オプションで追加の正規化を適用する。 ただ、これら副次的な機能は、voomの主要な目的と勘違いしてはいけない。 log-CPMへの標準化や正規化は他の関数で簡単にできるからである。
voomの主要な目的は、線形モデル内で使用される重みを計算することである。
発現変動を調べる
遺伝子ごとに線形モデルをfitする
vfit <- lmFit(v, design) vfit #An object of class "MArrayLM" #$coefficients # Basal LP ML laneL006 laneL008 #497097 2.583451 -4.873988 -4.340610 0.691359744 0.36149022 #27395 4.450225 3.966959 4.412685 -0.168486939 0.03518378 #18777 5.211960 4.918822 5.578919 0.008932751 0.20215237 #21399 5.236824 4.865704 4.828455 -0.022466033 0.16662215 #58175 -1.644309 3.843596 3.379532 -0.427881496 -0.28043183 #14160 more rows ... # #$stdev.unscaled # Basal LP ML laneL006 laneL008 #497097 0.2141729 0.6029739 0.5604689 0.2572328 0.7941610 #27395 0.1416249 0.1567280 0.1462306 0.1458866 0.2038254 #18777 0.1325387 0.1374684 0.1312922 0.1340760 0.1740921 #21399 0.1332442 0.1395319 0.1369334 0.1357618 0.1821041 #58175 0.3671817 0.1819939 0.1845398 0.2069245 0.2473389 #14160 more rows ... #(中略)
特定のコントラストについての統計計算
vfit <- contrasts.fit(vfit, contrasts = contr.matrix) vfit #An object of class "MArrayLM" #$coefficients # Contrasts # BasalvsLP BasalvsML LPvsML # 497097 7.4574387 6.92406052 -0.5333782 # 27395 0.4832662 0.03754016 -0.4457260 # 18777 0.2931380 -0.36695896 -0.6600970 # 21399 0.3711194 0.40836902 0.0372496 # 58175 -5.4879042 -5.02384089 0.4640633 #14160 more rows ... # #$stdev.unscaled # BasalvsLP BasalvsML LPvsML #497097 0.5664431 0.5269256 0.6319752 #27395 0.1714483 0.1650276 0.1645389 #18777 0.1548074 0.1512182 0.1458482 #21399 0.1564266 0.1548819 0.1499502 #58175 0.3494320 0.3498494 0.1987923 #14160 more rows ... #(中略)
次に、有意水準を計算する。 経験的ベイズアルゴリズムを使用して、 遺伝子固有の分散を全遺伝子の平均分散までに縮小する。
efit <- eBayes(vfit)
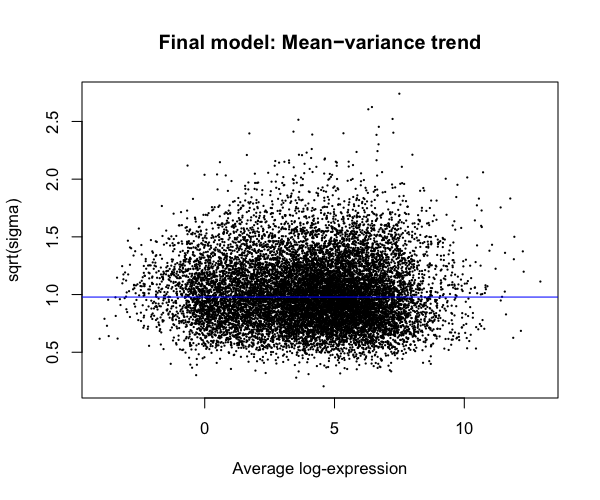
残余変動量と平均遺伝子発現量の診断プロットに見られるように、 voom関数は平均値と分散値の関係性をうまく取り除いた。
plotSA(efit, main = "Final model: Mean−variance trend") #quartz.save(file="Fig10.png", dpi=100, type="png", width=6, height=5)
ディスカッション・メモ
遺伝子間で情報を共有することの価値とは?
つまりは、遺伝子ごとに算出されたオリジナルの分散をなぜ利用しないのか?
結果を見る
結果の集計
summary(decideTests(efit)) # BasalvsLP BasalvsML LPvsML #Down 4127 4338 2895 #NotSig 5740 5655 8825 #Up 4298 4172 2445
効果量(effect size)の大きさが、次のDownstream解析で重要なら
(例えば,次なる実験のために遺伝子に優先順位をつけるとか)、
eBayes関数を使うよりも、
treat関数が提供するminimal log-fold-change法を選択する方が良いだろう。
tfit <- treat(vfit, lfc = 1) dt <- decideTests(tfit) summary(dt) # BasalvsLP BasalvsML LPvsML #Down 1417 1512 203 #NotSig 11030 10895 13780 #Up 1718 1758 182
ベン図を作成する
head(dt) #TestResults matrix # Contrasts # BasalvsLP BasalvsML LPvsML # 497097 1 1 0 # 27395 0 0 0 # 18777 0 0 0 # 21399 0 0 0 # 58175 -1 -1 0 # 108664 0 0 0 de.common <- BiocGenerics::which(dt[, 1] != 0 & dt[, 2] != 0) length(de.common) #[1] 2409 head(tfit$genes$SYMBOL[de.common], n = 20) # [1] "Xkr4" "Rgs20" "Cpa6" "Sulf1" "Eya1" "Msc" # [7] "Sbspon" "Pi15" "Crispld1" "Kcnq5" "Ptpn18" "Arhgef4" #[13] "Cracdl" "Aff3" "Npas2" "Tbc1d8" "Creg2" "Il1r1" #[19] "Il18r1" "Il18rap" vennDiagram(dt[, 1:2], circle.col = c("turquoise", "salmon")) #quartz.save(file="Fig11.png", dpi=100, type="png", width=6, height=5) write.fit(tfit, dt, file = "results.txt")

BiocGenerics::whichは、ベクトル、配列、またはリストのようなオブジェクトの中で、 TRUEとみなされる値のインデックスを返す。
上位発現変動の遺伝子を特定する
basal.vs.lp <- topTreat(tfit, coef = 1, n = Inf) basal.vs.ml <- topTreat(tfit, coef = 2, n = Inf) head(basal.vs.lp) # ENTREZID SYMBOL TXCHROM logFC AveExpr t P.Value adj.P.Val #12759 12759 Clu chr14 -5.442877 8.857907 -33.44429 3.990899e-10 2.703871e-06 #53624 53624 Cldn7 chr11 -5.514605 6.296762 -32.94533 4.503694e-10 2.703871e-06 #242505 242505 Rasef chr4 -5.921741 5.119585 -31.77625 6.063249e-10 2.703871e-06 #67451 67451 Pkp2 chr16 -5.724823 4.420495 -30.65370 8.010456e-10 2.703871e-06 #228543 228543 Rhov chr2 -6.253427 5.486640 -29.46244 1.112729e-09 2.703871e-06 #70350 70350 Basp1 chr15 -6.073297 5.248349 -28.64890 1.380545e-09 2.703871e-06 head(basal.vs.ml) # ENTREZID SYMBOL TXCHROM logFC AveExpr t P.Value adj.P.Val #242505 242505 Rasef chr4 -6.510470 5.119585 -35.49093 2.573575e-10 1.915485e-06 #53624 53624 Cldn7 chr11 -5.469160 6.296762 -32.52520 4.978446e-10 1.915485e-06 #12521 12521 Cd82 chr2 -4.667737 7.070963 -31.82187 5.796191e-10 1.915485e-06 #71740 71740 Nectin4 chr1 -5.556046 5.166292 -31.29987 6.760578e-10 1.915485e-06 #20661 20661 Sort1 chr3 -4.908119 6.705784 -31.23083 6.761331e-10 1.915485e-06 #15375 15375 Foxa1 chr12 -5.753884 5.625064 -28.34612 1.487280e-09 2.281914e-06
変動遺伝子を可視化する
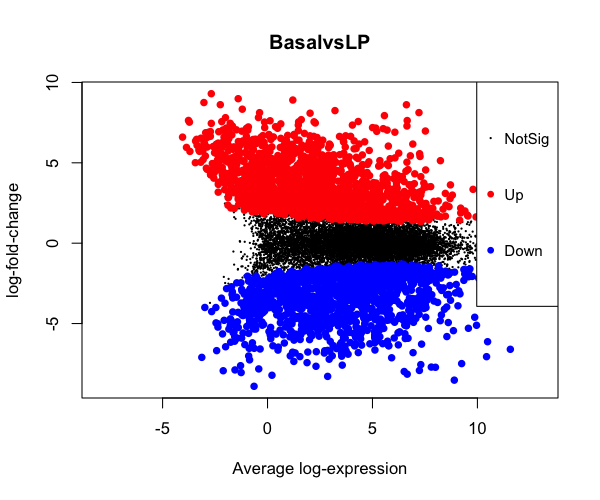
発現データの平均-差分プロット
limma::plotMD(tfit, column = 1, status = dt[, 1], main = colnames(tfit)[1], xlim = c(-8, 13)) #quartz.save(file="Fig12.png", dpi=100, type="png", width=6, height=5)
#Interactive plot Glimma::glMDPlot(tfit, coef = 1, status = dt, main = colnames(tfit)[1], side.main = "ENTREZID", counts = x1$counts, groups = group, launch = TRUE) #Fig13.png
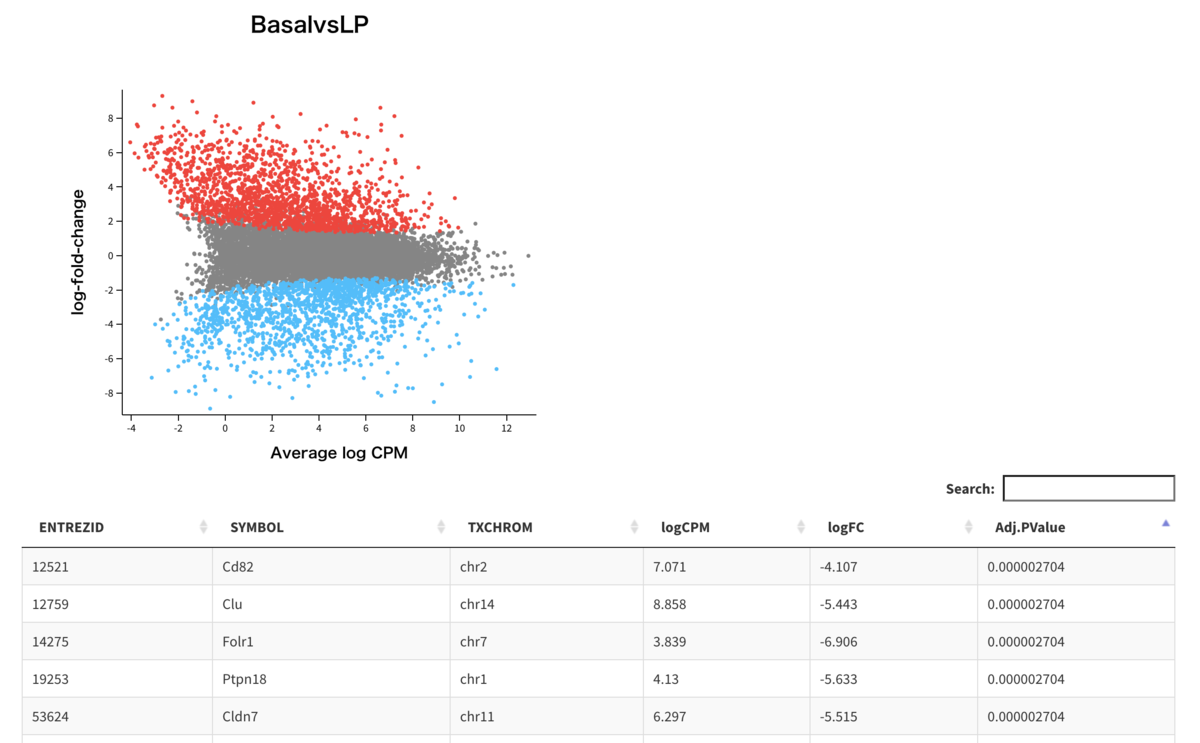
Basal細胞とLP細胞の発現変動遺伝子トップ100をヒートマップ表示
library("gplots") basal.vs.lp.topgenes <- basal.vs.lp$ENTREZID[1:100] i <- which(v$genes$ENTREZID %in% basal.vs.lp.topgenes) mycol <- colorpanel(1000, "blue", "white", "red") heatmap.2(v$E[i, ], scale = "row", labRow = v$genes$SYMBOL[i], labCol = group, col = mycol, trace = "none", density.info = "none") #quartz.save(file="Fig14.png", dpi=200, type="png", width=6, height=15)
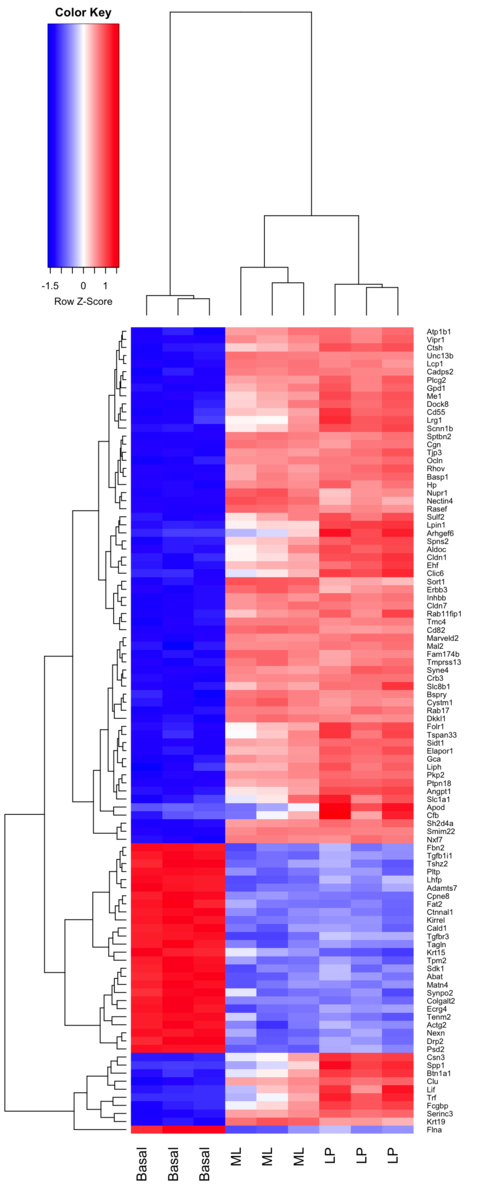
ディスカッション・メモ
ヒートマップでは、なぜ行単位(遺伝子単位)でスケーリングしたのか?
遺伝子セットのエンリッチメント評価
発現変動解析後に行う、一般的な最初のステップは、 特定の遺伝子セット(例えば、Gene Ontology(GO)カテゴリー、KEGGパスウェイ)のエンリッチメントを評価することである。 このチュートリアルでは、Broad Instituteが提供する遺伝子セットコレクション「MSigDB c2」を使用する。
#ロード load(system.file("extdata", "mouse_c2_v5p1.rda", package = "RNAseq123")) class(Mm.c2) #[1] "list" head(Mm.c2$KEGG_GLYCOLYSIS_GLUCONEOGENESIS) #[1] "100042025" "100042427" "103988" "106557" "109785" #[6] "110695" #遺伝子の識別子を遺伝子セットのインデックスに変換する idx <- limma::ids2indices(Mm.c2, id = rownames(v))
limmaのcamera関数は、すべての遺伝子セットを互いに比較することで、
特定のコントラストに対するエンリッチメントを評価する。
cam.BasalvsLP <- limma::camera(v, idx, design, contrast = contr.matrix[, 1]) head(cam.BasalvsLP, 5) # NGenes Direction PValue FDR #LIM_MAMMARY_STEM_CELL_UP 739 Up 1.134757e-18 5.360590e-15 #LIM_MAMMARY_STEM_CELL_DN 630 Down 1.569957e-15 3.708238e-12 #ROSTY_CERVICAL_CANCER_PROLIFERATION_CLUSTER 163 Up 1.437987e-13 2.264351e-10 #SOTIRIOU_BREAST_CANCER_GRADE_1_VS_3_UP 183 Up 2.181862e-13 2.576779e-10 #LIM_MAMMARY_LUMINAL_PROGENITOR_UP 87 Down 6.734613e-13 6.362863e-10 cam.BasalvsML <- limma::camera(v, idx, design, contrast = contr.matrix[, 2]) head(cam.BasalvsML, 5) # NGenes Direction PValue FDR #LIM_MAMMARY_STEM_CELL_UP 739 Up 5.090937e-23 2.404959e-19 #LIM_MAMMARY_STEM_CELL_DN 630 Down 5.132446e-19 1.212284e-15 #LIM_MAMMARY_LUMINAL_MATURE_DN 166 Up 8.875174e-16 1.397544e-12 #LIM_MAMMARY_LUMINAL_MATURE_UP 180 Down 6.287301e-13 7.425303e-10 #ROSTY_CERVICAL_CANCER_PROLIFERATION_CLUSTER 163 Up 1.684323e-12 1.591348e-09 cam.LPvsML <- limma::camera(v, idx, design, contrast = contr.matrix[, 3]) head(cam.LPvsML, 5) # NGenes Direction PValue FDR #LIM_MAMMARY_LUMINAL_MATURE_UP 180 Down 8.497295e-14 3.401020e-10 #LIM_MAMMARY_LUMINAL_MATURE_DN 166 Up 1.439890e-13 3.401020e-10 #LIM_MAMMARY_LUMINAL_PROGENITOR_UP 87 Up 3.840915e-11 6.048160e-08 #REACTOME_RESPIRATORY_ELECTRON_TRANSPORT 91 Down 2.655349e-08 3.135967e-05 #NABA_CORE_MATRISOME 222 Up 4.430361e-08 4.185805e-05
遺伝子セットのエンリッチメントをバーコードプロットで可視化する
limma::barcodeplot関数は、 ランク付けされた遺伝子リストにおいて、 1つまたは2つの遺伝子セットのエンリッチメントを表示する。
limma::barcodeplot(efit$t[, 3], index = idx$LIM_MAMMARY_LUMINAL_MATURE_UP, index2 = idx$LIM_MAMMARY_LUMINAL_MATURE_DN, main = "LPvsML") #quartz.save(file="Fig15.png", dpi=100, type="png", width=6, height=5)
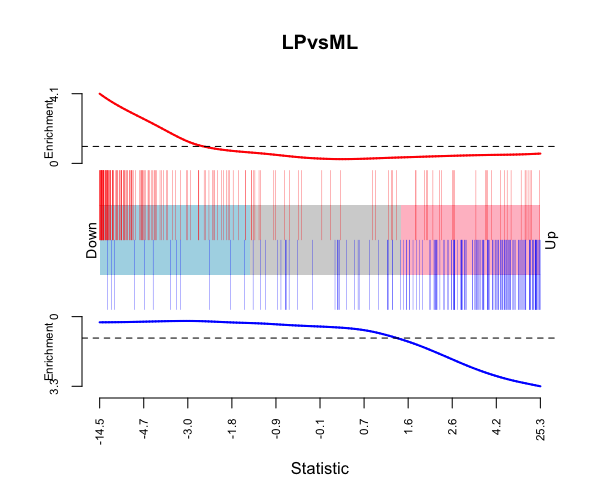
なお、オリジナルの分析では、ML-LPの対比で評価していたのに対して、 今回、LP-MLの対比で評価したために、 検定・変動の方向性はオリジナルの研究結果とは逆になっている。
生命科学データ・RNA-seq解析についての関連図書
RNA-seq解析を学ぶにあたって、R言語の基本的な使い方であったり、 バイオインフォの基本的な知識にふれておくのが良いと思いますので、 関連図書を以下にあげておきます。ご参考までに。
参考資料
関連記事
# The limma User's Guide (run `limmaUsersGuide()` in the R console) is # very useful. Especially see Section 2.1 for citations to the primary # publications that describe the methods and Chapter 9 for how to # contruct the model for different types of study designs. # The Bioconductor support site (https://support.bioconductor.org/) # has years worth of questions and answers about RNA-seq analysis and # other topics in bioinformatics. # Overview of RNA-seq analysis # # A. Oshlack, M. D. Robinson and M. D. Young. “From RNA-seq reads to # differential expression results”. In: _Genome Biology_ 11.12 (2010), # p. 220. DOI: 10.1186/gb-2010-11-12-220. # R/Bioconductor tutorial starting from fastq files # # Chen Y, Lun ATL and Smyth GK. From reads to genes to pathways: # differential expression analysis of RNA-Seq experiments using # Rsubread and the edgeR quasi-likelihood pipeline [version 2; # referees: 5 approved]. F1000Research 2016, 5:1438 (doi: # 10.12688/f1000research.8987.2) # Comparisons of RNA-seq methods for differential expression testing # # F. Rapaport, R. Khanin, Y. Liang, et al. “Comprehensive evaluation # of differential gene expression analysis methods for RNA-seq data”. # In: _Genome Biology_ 14.9 (2013), p. R95. DOI: # 10.1186/gb-2013-14-9-r95. # # C. Soneson and M. Delorenzi. “A comparison of methods for # differential expression analysis of RNA-seq data”. In: _BMC # Bioinformatics_ 14.1 (2013), p. 91. DOI: 10.1186/1471-2105-14-91. # limma # # Ritchie ME, Phipson B, Wu D, et al.: limma powers differential # expression analyses for RNA-sequencing and microarray studies. # Nucleic Acids Res. 2015; 43(7): e47. # Glimma # # Su S, Ritchie ME: Glimma: Interactive HTML graphics for RNA-seq # data. 2016; R package version 1.1.1. # edgeR # # Robinson MD, McCarthy DJ, Smyth GK: edgeR: a Bioconductor package # for differential expression analysis of digital gene expression # data. Bioinformatics. 2010; 26(1): 139–140. # Bioconductor project # # Huber W, Carey VJ, Gentleman R, et al.: Orchestrating # high-throughput genomic analysis with Bioconductor. Nat Methods. # 2015; 12(2): 115–121. # TMM normalization # # Robinson MD, Oshlack A: A scaling normalization method for # differential expression analysis of RNA-seq data. Genome Biol. 2010; # 11(3): R25. # limma+voom # # Law CW, Chen Y, Shi W, et al.: voom: Precision weights unlock linear # model analysis tools for RNA-seq read counts. Genome Biol. 2014; # 15(2): R29 # Empirical Bayes to estimate gene expression variance # # Smyth GK: Linear models and empirical bayes methods for assessing # differential expression in microarray experiments. Stat Appl Genet # Mol Biol. 2004; 3(1): Article3.