
- はじめに
- 関連パッケージの読み込み
- SRA Toolkit / fastq-dump コマンドを使って、FASTQファイルを取得する。
- FASTQファイルの読み込み
- リード長を取り出す方法
- クオリティスコアを可視化する
- サイクルごとにクオリティスコアを可視化する
- まとめ
はじめに
今回は、MacOSX環境で、SRA Toolkitのfastq-dumpを用いて取得した、 FASTQ形式ファイルをRに読み込む方法を扱う。
FASTQ形式ファイルの読み込みには、
Bioconductorパッケージの1つである、
ShortReadパッケージのreadFastq関数を用いる。
readFastq関数は、
指定のディレクトリ内にあるFASTQ形式のファイルのうち、
ファイル名がパターンマッチするものをすべて読み込む。
あるいは、1つのファイル名を与えて、読み込むんこともできる。
読み込み結果は、ファイル内の配列と品質スコアをコンパクトな内部表現として返す。
それでは、早速、FASTQファイルの読み込みを試してみる。
関連パッケージの読み込み
#インストール install.packages(c("BiocManager", "ggplot2")) BiocManager::install("ShortRead") #ロード library(ShortRead) library(ggplot2)
SRA Toolkit / fastq-dump コマンドを使って、FASTQファイルを取得する。
SRA Toolkitの設定は、以前の記事を参考のこと。
ターミナルあるいはRStudioのTerminalタブにて、以下を実行する。
# fastq-dumpのパス確認 which fastq-dump #SRR17327096データの取得(約16MB) fastq-dump SRR17327096 #Read 110774 spots for SRR17327096 #Written 110774 spots for SRR17327096 #ファイルの確認 ls | grep SRR17327096 #SRR17327096.fastq
FASTQファイルの読み込み
それでは、readFastq関数で、
FASTQファイルを読み込む。
#読み込み fq <- ShortRead::readFastq("SRR17327096.fastq") #表示 fq #class: ShortReadQ #length: 110774 reads; width: 488 cycles #シークエンスをみる reads <- ShortRead::sread(fq) reads
リード長を取り出す方法
#リード長を取り出す widths <- as.data.frame(reads@ranges@width) #head表示 head(widths) # reads@ranges@width #1 488 #2 488 #3 488 #4 488 #5 488 #6 488 #カウント table(widths) #widths # 488 #110774
クオリティスコアを可視化する
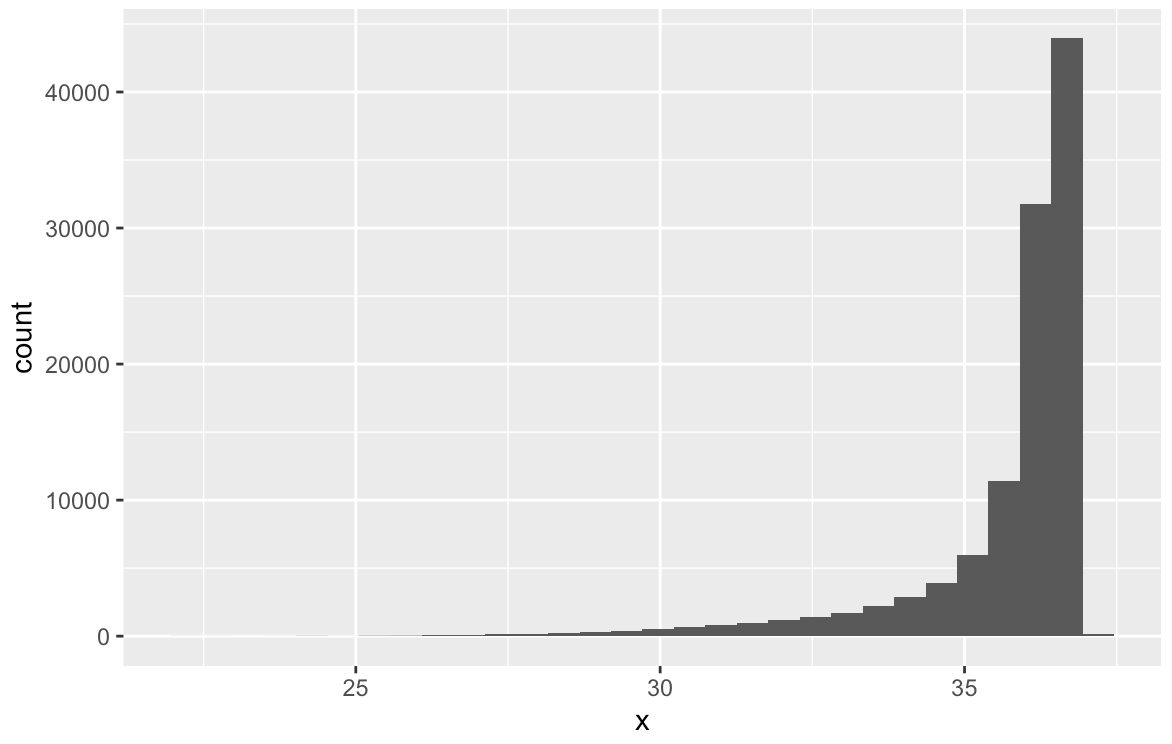
ここでは、FASTQファイルのクオリティスコアを抽出して、リードごとの平均値をグラフ化する。
# クオリティスコアを得る quals <- Biostrings::quality(fq) #表示 quals #class: FastqQuality #quality: #BStringSet object of length 110774: # width seq # [1] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FFFFFFFFF:FFFFFF,FFFFFFFFFF: # [2] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FFFFFFFFFFFFFFFFF:FFFFFF:FFF # [3] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...,FFFFFFFFFFFFFFFFFFF:FF,FFFF # [4] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FF:FFFF:FFFFF,FF::FFFFFFF,FF # [5] 488 FFFFFFFFFFFFFFFFFFFFFFF:FFFFF...FFFFFFFFF,FFFFFFF:F,FFFFFFFF # ... ... ... #[110770] 488 FFFFFFFFFFFFFFFFFFFFFFFFFF:FF...FFFF:FFF,FFFFFFFFFF:FFFFF:FF #[110771] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FFFFFFFFFFFFFFFFFFFFFFFFFFFF #[110772] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FFFFFFFFFFFFFFFFFFF:FFFF:FFF #[110773] 488 FFFFFFFFFFFFFFFFFFFFFFFFF:FFF...F:FFFFFFFFFFFFFF:FFFFFFFFFF: #[110774] 488 FFFFFFFFFFFFFFFFFFFFFFFFFFFFF...FFFFFFFFFFFFFFFFFFFFFFFFFFFF #オブジェクトを強制的に行列クラスに変換する scores <- as(quals, 'matrix') #行ごとで平均値を計算 Avgscore <- data.frame(x=rowMeans(scores, na.rm = T)) str(Avgscore) #'data.frame': 110774 obs. of 1 variable: # $ x: num 36.4 36.8 36.3 36.5 35.8 ... #可視化 ggplot(Avgscore) + geom_histogram(aes(x=x))
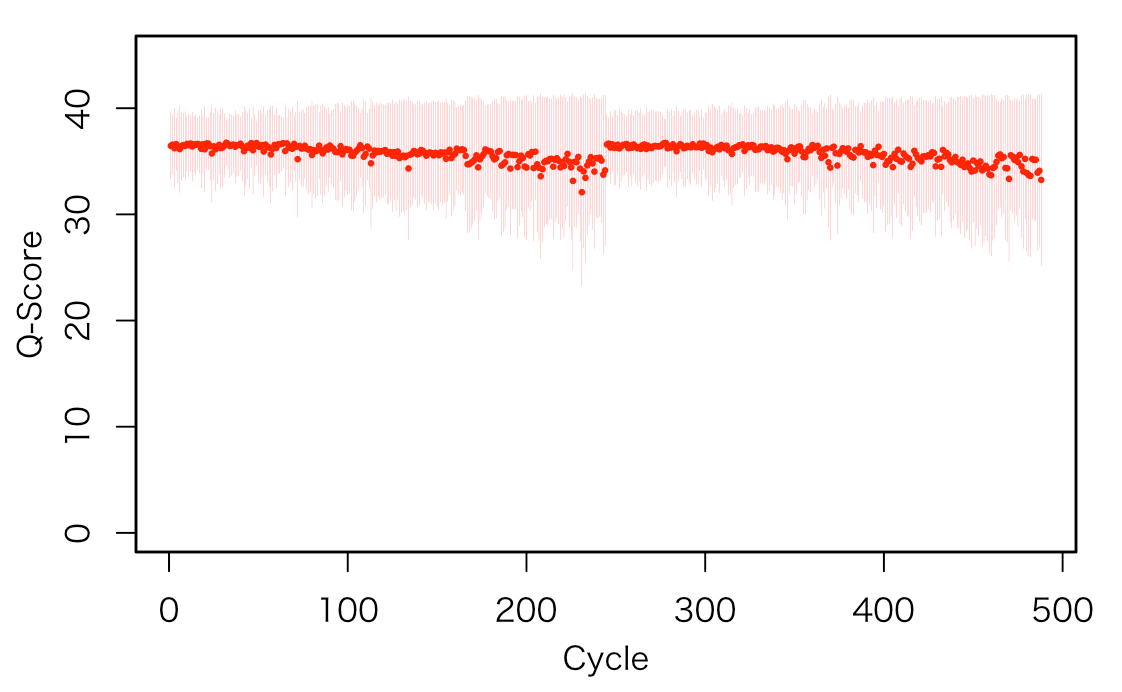
サイクルごとにクオリティスコアを可視化する
#リストに変換する Dat <- NULL for(n in 1:ncol(scores)){ #n <- 1 print(n) a <- mean(scores[,n]) b <- sd(scores[,n]) cc <- data.frame(Ave=a, sd=b) Dat <- rbind(Dat, cc) } str(Dat) #'data.frame': 488 obs. of 2 variables: # $ Ave: num 36.5 36.6 36.2 36.6 36.3 ... # $ sd : num 3.17 2.8 3.79 2.5 3.4 ... #可視化 par(family="HiraKakuProN-W3", lwd=1.5, xpd=F, mgp=c(2.25, 1, 0), mai=c(0.75,0.75, 0.5, 0.5)) plot(c(1, nrow(Dat)), c(0, 45), type = "n", xlab="Cycle", ylab="Q-Score") points(Dat[,1], pch=21, col = "red", cex=0.25) for(m in 1:ncol(scores)){segments(m, Dat[m,1]+Dat[m,2], m, Dat[m,1]-Dat[m,2], col = "red", lwd=0.1) }
まとめ
NCBIアーカイブからFASTQファイルのダウンロードから、 ファイル読み込みまでの内容を扱った。
Bioconductorプロジェクトから関数群が提供されていることにも少し感動する。