
- はじめに
- Open-Babelの基本動作
- PubChemからの化合物の情報・構造データの取得
- Rで、PubChem化合物データを読み込み
- 「gluc」あるいは「glyc」を含む化合物名を検索する
- ImageMagickを用いて、ヒットした化合物の2D画像を作成する
- 検索された化合物構造(sdf)をPyMolで可視化する
- まとめ
- 関連資料
はじめに
情報学と化学(バケガク)との融合領域を、「ケモインフォマティクス」というらしいです。略して、「ケモインフォ」です。
この用語の由来はchemistryとinformaticsとを合わせた造語で、
私はあまり使ったことないですが、日本語では「情報化学」とも言われるようです。
今回、タンパク質とリガンドの結合予測を行う、ドッキングシミュレーションの下準備として、 PubChemからの化合物情報の取得、Open Babelを使った化合物構造の作成やフォーマット変換、PyMOLを使った構造可視化といった諸々についてまとめてみました。
Open Babelは、主に化学構造ファイルフォーマットを変換するために用いられるフリーソフトウェアで、化学構造ファイルの作成やエネルギーに基づく構造の最適化計算などを行えます。また、PyMOLは老舗のオープンソースの分子グラフィックスツールで、現在、Schrödinger社によって商品化されて提供されています。
では、始めてみましょう。
実行環境
実行環境は以下の通りです。
macOS Big Sur (バージョン11.5.2) MacBook Air (M1, 2020) チップ Apple M1 メモリ 16GB
Rパッケージの読み込み
R環境を立ち上げて、必要なパッケージをインストールしておきます。今回は少なめです。
#パッケージ・インストール pack <- c("readr") install.packages(pack[!(pack %in% unique(rownames(installed.packages())))]) #ロード for(n in 1:length(pack)){ eval(parse(text = paste0("library(", pack[n], ")"))) }; rm("n", "pack")
Open-Babel & PyMolのインストールについて
Macの場合、HomebrewでOpen-BabelとPyMolをインストールするのが便利です。
まずは、ターミナルを起動して、open-babelをbrewコマンドでインストールします。
#Open-Babel のインストール brew install open-babel #パス確認 which obabel #/opt/homebrew/bin/obabel #ヘルプ表示 obabel -H #バージョン確認 obabel -V #Open Babel 3.1.0 -- Aug 5 2022 -- 07:11:30
Open-Babelをインストールすると、obabel、obconformer、obdistgen、obenergy、obfitなど、19個のコマンド群がインストールされます。主には、obabelを使います*1。
次に、pymolをインストールします。現在、PyMOL(TM) Molecular Graphics System, Version 2.5.0.が使えます。
#pymolのインストール brew install pymol #パス確認 which pymol #/opt/homebrew/bin/pymol
これで、open-babelとpymolの設定は完了です。
また、画像結合時に使うので、ImageMagickもインストールしておきます。
#ImageMagickのインストール
brew install imagemagick
Open-Babelの基本動作

ここでは、Open-Babelの基本的な使い方について解説します。 まずは、SMILES記法をスタートに、化学構造ファイルを作成する方法を紹介します。
SMILES形式は多くの種類の分子エディタにおいてもインポート可能で、二次元の図表あるいは三次元のモデルとして表示できます。また、CANGENアルゴリズムに基づいて1つの表記になるよう正規化された(canonical)ものを、カノニカルSMILESと呼ばれます。
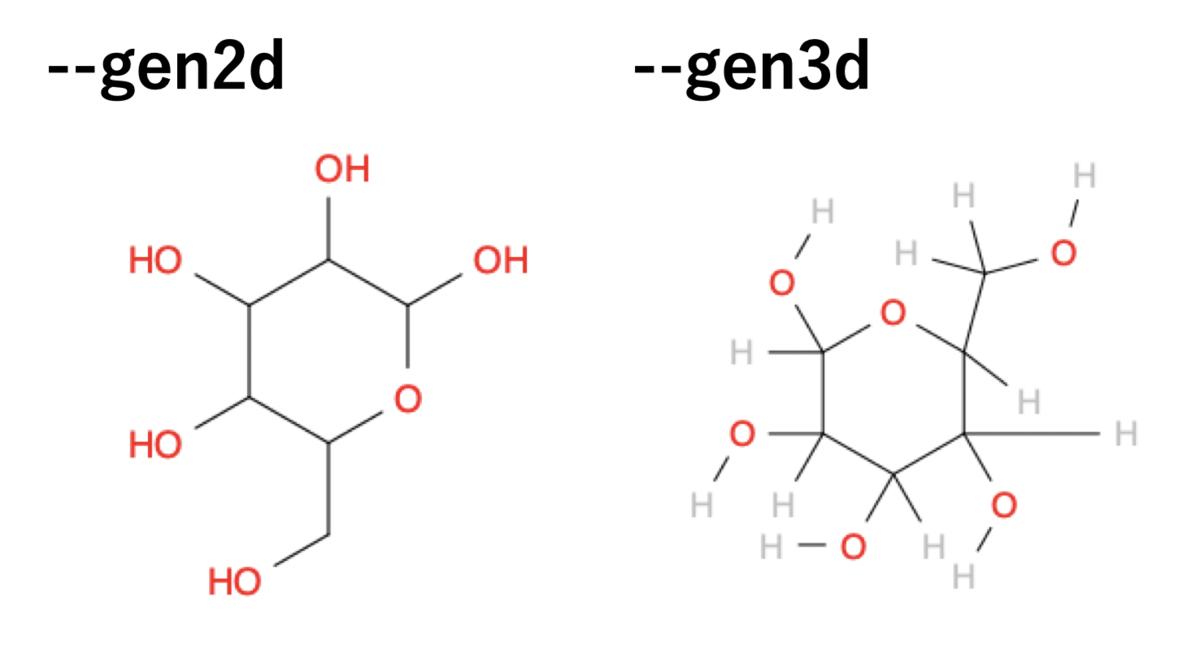
実施例として、グルコースのSMILES形式を使って、2Dと3Dの構造ファイルを作成してみます。 グルコースのCanonical SMILESは、「C(C1C(C(C(C(O1)O)O)O)O)O」と記述されます。
まずは、obabelコマンドを使って、グルコースのSMILESを2D画像(.png)に変換します。SMILESの入力については、-:"<SMILES string>" or -:'<SMILES string>'と記述します。また、別記法として、smiファイルを使う方法、echoを使う方法も記載しています。
それでは、ターミナルでコマンドを実行していきます。
#SMILESを2D画像に変換 + --gen2D obabel -:'C(C1C(C(C(C(O1)O)O)O)O)O' -o png -O out1.png --gen2D #SMILESを3D画像に変換 + --gen3D obabel -:'C(C1C(C(C(C(O1)O)O)O)O)O' -o png -O out2.png --gen3D #別記法(1): smiファイルを使う場合 echo 'C(C1C(C(C(C(O1)O)O)O)O)O' > glucose.smi head glucose.smi #C(C1C(C(C(C(O1)O)O)O)O)O obabel -i smi glucose.smi -o png -O out3.png --gen3D #別記法(2): echoを使う #SMILESを2D画像に変換 echo 'C(C1C(C(C(C(O1)O)O)O)O)O' | obabel -i smi -o png -O out4.png --gen2d #SMILESを3D画像に変換 echo 'C(C1C(C(C(C(O1)O)O)O)O)O' | obabel -i smi -o png -O out5.png --gen3D
次に、SMILESをPDB/PDBQT形式に変換します。
#SMILESをPDB形式に変換 obabel -:'C(C1C(C(C(C(O1)O)O)O)O)O' -o pdb -O glucose.pdb --gen3D #SMILESをPDBQT形式に変換 obabel -:'C(C1C(C(C(C(O1)O)O)O)O)O' -o pdbqt -O glucose.pdbqt --gen3D #SMILESをmol2形式に変換 obabel -:'C(C1C(C(C(C(O1)O)O)O)O)O' -o mol2 -O glucose.mol2 --gen3D #pymolでPDBファイルを可視化 pymol glucose.pdb
pymolで開くと、glucoseの3D構造が下図にように見えます。

Open-Babelの関連コマンドについて
続いて、Open-Babelの関連コマンドについて紹介します。
まず、obenergyコマンドは、分子のエネルギーを計算します。
#GAFFによるエネルギー計算 obenergy -ff GAFF -v glucose.pdb #... #E N E R G Y # # TOTAL BOND STRETCHING ENERGY = 2.055 kJ/mol # TOTAL ANGLE BENDING ENERGY = 15.645 kJ/mol # TOTAL TORSIONAL ENERGY = 9.809 kJ/mol # TOTAL IMPROPER-TORSIONAL ENERGY = 0.000 kJ/mol # TOTAL VAN DER WAALS ENERGY = 31.348 kJ/mol # TOTAL ELECTROSTATIC ENERGY = -7.801 kJ/mol # #TOTAL ENERGY = 51.056 kJ/mol
obpropコマンドは、標準的な分子特性を出力します。
obprop glucose.pdb #name glucose.pdb 1 #formula C6H12O6 #mol_weight 180.156 #exact_mass 180.063 #canonical_SMILES OC[C@H]1O[C@H](O)[C@@H]([C@@H]([C@@H]1O)O)O # #InChI InChI=1S/C6H12O6/c7-1-2-3(8)4(9)5(10)6(11)12-2/h2-11H,1H2/t2-,3-,4-,5-,6+/m1/s1 # #num_atoms 24 #num_bonds 24 #num_residues 1 #num_rotors 1 #sequence UNL #num_rings 1 #logP -3.2214 #PSA 110.38 #MR 35.736 #$$$$
obminimizeコマンドは、形状を最適化し、分子のエネルギーを最小になるように計算します。
また、obconformerコマンドは、低エネルギーコンフォーマーを生成します。
#GAFFによる構造 & エネルギー最適化: 2500計算がdefault obminimize -n 2500 -ff GAFF glucose.pdb > glucose_obm.pdb #pymolで可視化 pymol glucose.pdb glucose_obm.pdb #Monte Carlo searchによりランダム構造を作製した後に最も低いエネルギー状態を選択 #250個のランダム構造の生成、100 stepsの構造最適化 obconformer 250 100 glucose.pdb > glucose_obc.pdb #pymolで可視化 pymol glucose.pdb glucose_obc.pdb

計算前後の結果を重ね合わせていますが、ほとんど構造は変わってませんね。
PubChemからの化合物の情報・構造データの取得
PubChemは、国立生物工学情報センター(NCBI)によって維持管理されている、化学分子データベースの1つです。化合物情報の収録数は、確実に、世界一位でしょうね。
今回、PubChemから、「薬剤・薬物情報」に関する化合物の情報や構造をダウンロードします。 それでは、PubChemのWebページに訪問します。
これが、PubChemのホーム画面です。そこで、検索窓の下にある、「Browse Data」に進みます。
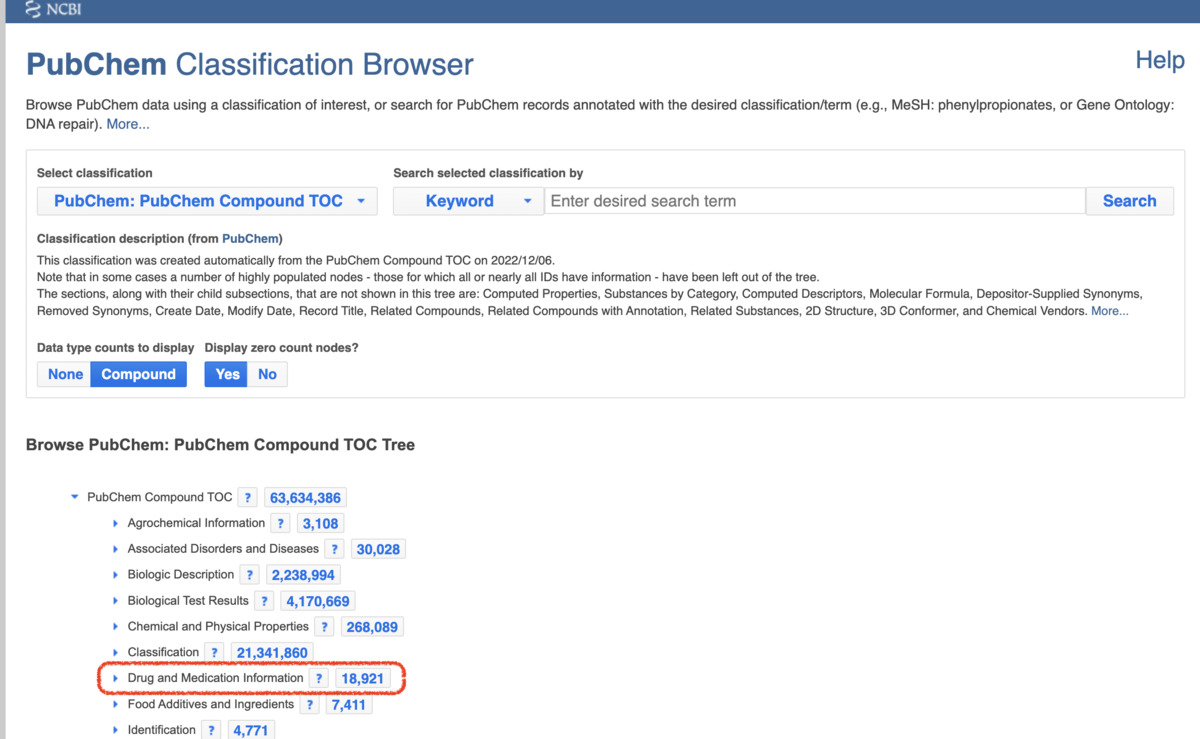
「薬剤・薬物情報」のデータを収集するため、「Drug and Medication Information」を選択します。 各自、必要に応じて、別カテゴリーを選択してください。化学物質数としては、約1.8万種類が収録されています。
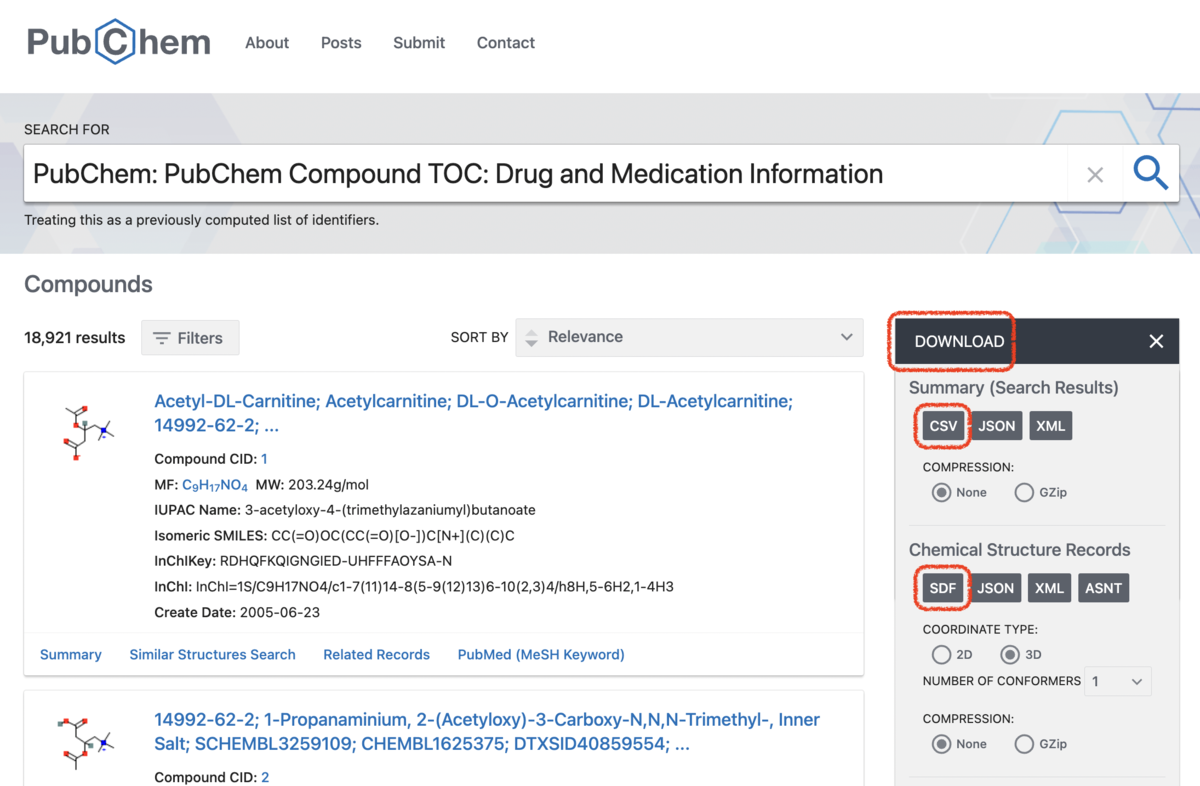
続いて、右側のダウンロードのタブをクリックして、csv形式とsdf形式のファイルをそれぞれダウンロードします。ダウンロード後に、PubChem_Drug_Medication_Information.csv、PubChem_Drug_Medication_Information.sdfという名前をつけて、作業ディレクトリ内に保存しました。
Rで、PubChem化合物データを読み込み
それでは、PubChemの化合物データ、csvファイルをR環境で読み込みます。 ここでは、readrパッケージのread_csvを使います。
#csv読み込み PubChem <- readr::read_csv("PubChem_Drug_Medication_Information.csv", show_col_types = FALSE) #表示 head(PubChem) # A tibble: 6 × 24 # cid cmpdname cmpds…¹ mw mf polar…² compl…³ xlogp # <dbl> <chr> <chr> <dbl> <chr> <dbl> <dbl> <chr> #1 1 Acetylca… Acetyl… 203. C9H1… 66.4 214 0.400 #2 2 1-Propan… 14992-… 204. C9H1… 63.6 219 -0.3… #3 6 1-Chloro… 1-chlo… 203. C6H3… 91.6 224 2.300 #4 11 1,2-Dich… 1,2-di… 99.0 C2H4… 0 6 1.500 #5 34 2-Chloro… 2-chlo… 80.5 C2H5… 20.2 10 -0.1… #6 38 2-Dehydr… 2-dehy… 146. C6H1… 74.6 159 -0.1… # … with 16 more variables: heavycnt <dbl>, # hbonddonor <dbl>, hbondacc <dbl>, rotbonds <dbl>, # inchi <chr>, isosmiles <chr>, inchikey <chr>, # iupacname <chr>, meshheadings <chr>, annothits <chr>, # annothitcnt <dbl>, aids <chr>, cidcdate <dbl>, # sidsrcname <chr>, depcatg <chr>, annotation <chr>, and # abbreviated variable names ¹<200b>cmpdsynonym, ²<200b>polararea, … # ℹ Use `colnames()` to see all variable names #列名 colnames(PubChem) # [1] "cid" "cmpdname" "cmpdsynonym" # [4] "mw" "mf" "polararea" # [7] "complexity" "xlogp" "heavycnt" #[10] "hbonddonor" "hbondacc" "rotbonds" #[13] "inchi" "isosmiles" "inchikey" #[16] "iupacname" "meshheadings" "annothits" #[19] "annothitcnt" "aids" "cidcdate" #[22] "sidsrcname" "depcatg" "annotation"
1列目にcid、2列目にcmpdname、3列目にcmpdsynonym、8列目にxlogp、14列目にisosmiles、などの情報が含まれています。
「gluc」あるいは「glyc」を含む化合物名を検索する
glucoseの一部の「gluc」あるいは「glyc」を使って、3列目のcmpdsynonym内を検索して、化合物を探します。
#「gluc|glyc」でcmpdsynonym列を検索 PubChem0 <- PubChem[grepl("gluc|glyc", PubChem$cmpdsynonym),] #cmpdname表示 head(PubChem0$cmpdname) #[1] "2-Chloroethanol" "Ethylene Glycol" "Betaine" "Trimethyl glycine" #[5] "2,3-Butanediol" "Cellobiose" #行列数 dim(PubChem0) #[1] 1361 24
「gluc」or「glyc」で検索した結果、1361個の化合物がヒットしました。
ImageMagickを用いて、ヒットした化合物の2D画像を作成する
ここでは、ヒットした1361個の化合物のSMILESをもとに、2D画像(PNG)に変換します。
#SMILES情報 smiles <- PubChem0$isosmiles #表示 head(smiles) #[1] "C(CCl)O" #[2] "C(CO)O" #[3] "C[N+](C)(C)CC(=O)[O-]" #[4] "C[N+](C)(C)CC(=O)O" #[5] "CC(C(C)O)O" #[6] "C(C1C(C(C(C(O1)OC2C(OC(C(C2O)O)O)CO)O)O)O)O" #2D画像に変換 for(n in 1:length(smiles)){ #n <- 1 a <- paste0("obabel -:'", smiles[n], "' -o png -O gluc_out_", formatC(n, width = 2, flag = "0"), ".png --gen2D") #実行 system(a) } #ImageMagickを使った画像結合 system("montage -tile 15x15 -geometry 100% gluc_out_*.png gluc_out.png")
以下が、作成した化合物構造の画像です。



検索された化合物構造(sdf)をPyMolで可視化する
"ついで"くらいの内容ですが、ターミナルを起動して、pymolコマンドでsdfファイルが開いてみます。
pymol PubChem_Drug_Medication_Information.sdf
まとめ
PubChemからの化合物の情報・構造の取得方法とか、Open-BabelやPyMolコマンドについての使い方とかについて解説しました。
この内容は、主に、ドッキングシミュレーション用の下準備ですので、次回に続きます。。。
関連資料
*1:以前は、babelだったような気がしますが、obabelに名前が変わったようですね。